Editor’s note: This text-based course is an edited transcript of the webinar, Cystic Fibrosis: A Comprehensive Review, presented by Prigi Varghese, RN, PNP.
Learning Outcomes
After this course, participants will be able to:
- Recognize the basic pathophysiology of Cystic Fibrosis
- Describe how to screen and diagnosis CF
- Describe how CF affects the body systems
- Identify treatments used in the management of CF
- Discuss the multidisciplinary approach to CF care
Introduction
I have been with the Cystic Fibrosis Center in Dallas for the past seven years and very excited to share an overview of Cystic Fibrosis. Respiratory therapists, please know that you guys are instrumental to the CF community. Thank you for the work that you do.
What Is Cystic Fibrosis?
Cystic Fibrosis is a progressive autosomal recessive genetic disorder that affects multiple body systems. The mutations or errors in the Cystic Fibrosis transmembrane conductance regulator gene (CFTR) disrupts the sodium chloride ion exchange within the cells (Figure 1), causing a buildup of thick mucus in the lungs, pancreas, and other organs.
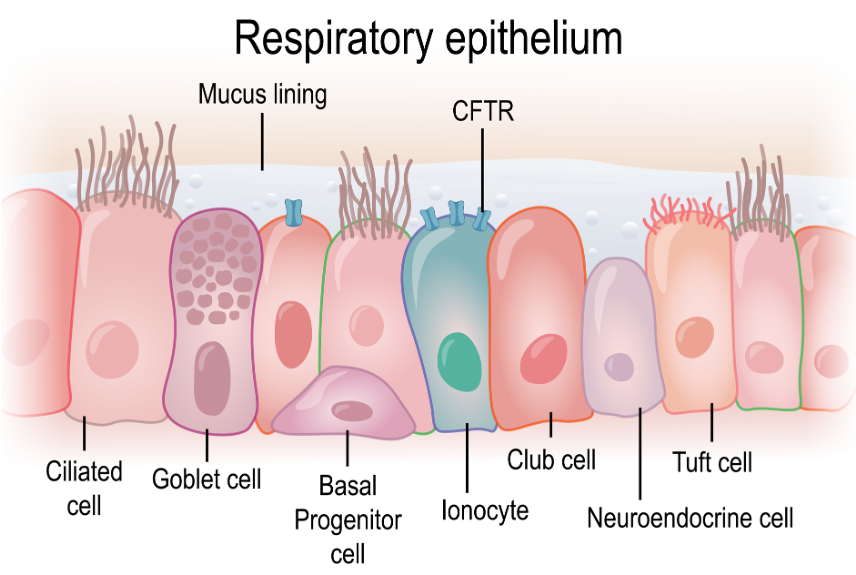
Figure 1. Illustration of the CFTR gene (Source: Continued licensed from Getty Images).
CFTR:
Cystic Fibrosis Transmembrane Conductance Regulator
The cystic fibrosis transmembrane conductance regulator (CFTR) gene located on chromosome number seven, contain codes from each parent to create the CFTR chloride protein channel within the cells. If the genes are without any mutations, the CFTR protein, as you can see in this diagram (Figure 2), can transport chloride in and out of the cells without any disruption. The CFTR gene was identified in 1989 and is responsible for encoding the CFTR protein, the CFTR protein regulates the chloride ion content of epithelial cells that line nasal cavities, lungs, and stomach to name a few. These cells secrete fluids such as sweat, mucus, and tears.
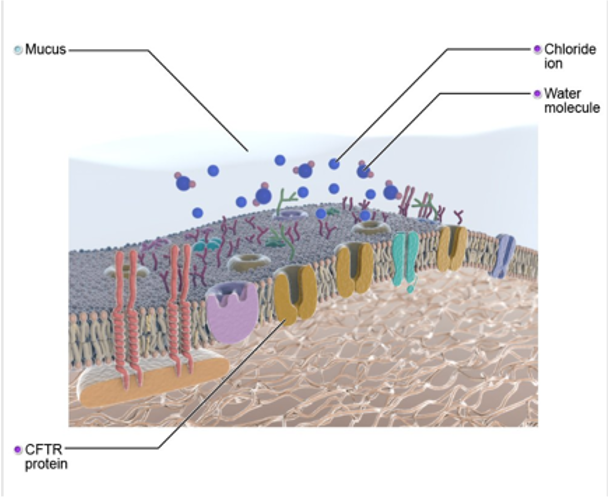
Figure 2. Diagram of CFTR gene (Source: Primal Images).
When there is a defect with the CFTR gene (Figure 3), as in the case of Cystic Fibrosis, the CFTR protein is not working correctly and is unable to help move chloride, a component of salt, to the surface. The mucus then on the cell surface becomes very thick and sticky.
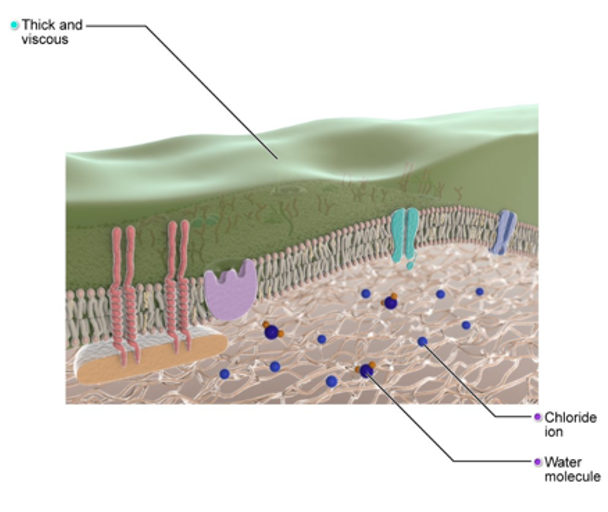
Figure 3. Illustration of the CFTR gene mutation (Source: Primal Images).
CFTR Mutation Class
There are over 2000 Cystic Fibrosis mutations that have been identified worldwide. Therefore, the scientists have grouped these mutations into five classes by the problems that they cause and the production of the CFTR protein. Class one is the protein production mutations, which interfere with the production of the CFTR protein. Class two is the protein processing mutations, and these mutations cause an amino acid to be deleted or incorrect amino acid to be added. Therefore, the CFTR protein cannot form its correct shape and function properly. Class three is the gating mutations. The CFTR protein is generally shaped like a tunnel or channel with a gate. The cell can open the gate when chloride needs to flow through the channel. Otherwise, the gate stays closed. Gating mutations lock the gate in the closed position so that chloride cannot get through at all. Class four is the conduction mutations, and these mutations change the shape of the inside of the tunnel so that chloride cannot move through as quickly as it should. Finally, class number five is the insufficient protein mutations that cause a reduction in the amount of normal CFTR protein at the cell surface, no matter what class the mutation is in at the end of the day. They all lead to disruption of sodium and chloride exchange in the cells. Sodium-ion plus chloride ion make up salt, and we know in the body, water follows salt.
The Gene Factor
The top diagram (Figure 4) shows a normal cell that does not have any defective CFTR genes. The exchange between sodium and chloride is balanced, and therefore the water can enter and exit quickly from the cell. In the bottom diagram (as shown in Figure 4), the CFTR gene is not there, or it is defective. This disrupts the sodium chloride exchange in the cell, causes the water to accumulate inside the cell and not disperse the cell's surface to help keep the mucus out. How are genes passed along? The diagram in Figure 4 shows mom and dad, and both of them are carriers for CF, meaning they each have one Cystic Fibrosis mutation. If both are carriers, then with each pregnancy, they have a 25% chance of having a child that has Cystic Fibrosis, a 25% chance of having a child without Cystic Fibrosis and not a carrier, and a 50% chance of having a child that is a CF carrier.
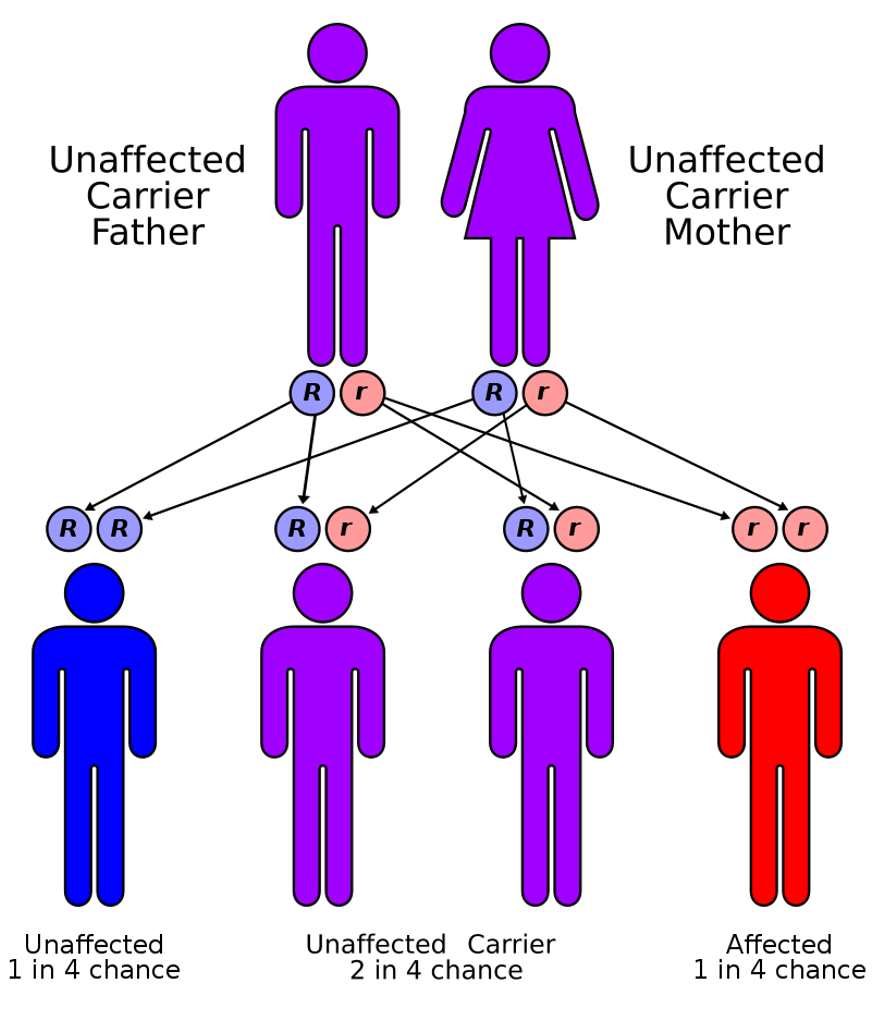
Figure 4. Diagram of the Cystic Fibrosis gene factor (Source: Autorecessive.svg CC BY 3.0 via Wikimedia Commons).
Cystic Fibrosis Stats
Here are a few current statistics on CF from the CF Foundation. The CF Foundation sets forth the evidence-based guidelines for every aspect of care, and all CF centers must adhere to these guidelines, or we can lose our accreditation and funding for our centers. There are over 100 CF centers across the nation, more than 30,000 people living in the United States currently have a diagnosis of Cystic Fibrosis, and worldwide there are over 70,000 people. Approximately a thousand new cases of CF are diagnosed every year. More than 75% of people with Cystic Fibrosis are diagnosed by two due to newborn screening. Right now, more than half the CF population is age 18 or older. Meaning our patients are living longer.
Race Distribution
Years ago, Cystic Fibrosis was thought to be a European or Caucasian disease. However, we see Cystic Fibrosis in all races and ethnicities. One in approximately 3,500 white newborns is diagnosed with CF, while one in 17,000 African-American babies and one in 31,000 Asian-American newborns. Recently, patients of Hispanic origin with Cystic Fibrosis are the largest growing minority representing about 8.5% of patients in the United States with Cystic Fibrosis.
SCREENING
Let's look at different types of screening for Cystic Fibrosis.
Prenatal Screening
CF can be screened prenatally. The American College of Obstetricians and Gynecologists recommended CFTR 32 mutation panel to screen expecting couples for CS carrier status. Especially if there was a family history on either side, the screen is more sensitive among the Caucasian population at about 78%. Sensitivity is lower in other races because they could have a mutation not listed on that 32 mutation panel.
Newborn Screen For Cystic Fibrosis
The most common and widely used is the newborn screen. A newborn screen is a simple heel stick done at birth. And then one to two weeks repeated at the pediatrician's office—the newborn screen checks over 53 genetic conditions. Cystic Fibrosis was added to this list as recently as 2009 in the state of Texas. This means that kids born before 2009 were not screened at birth for CF. Those affected are now being picked up on clinical signs and symptoms by their pediatricians or other specialty doctors like pulmonologists, allergists, GI doctors, and ENT doctors.
Specific to CF, the newborn screen checks for a chemical called immunoreactive trypsinogen (IRT). IRT is made by the pancreas and helps with digestion. Everyone has small amounts in their body, but IRT can be high or very elevated in infants with Cystic Fibrosis. If the IRT is high, the second step is to look for changes in the gene that causes CF. Newborn screen checks for the most common mutations in each state. It can vary between states. In Texas, our newborn screen only has 60 of the most common mutations, but as mentioned before, there are over 2,000 mutations documented. This is why we advise pediatricians and families to obtain a sweat test.
How Is CF Diagnosed?
With increased diversity in all of our states, there is a high chance a patient could have a mutation that is not on the newborn screen mutation panel. But not all elevated IRT means the baby has CF. Premature birth and or traumatic delivery can stress out the pancreas, causing it to release a high amount of IRT into the bloodstream. This is why in Texas, we wait for the second newborn screen. If the IRT is also elevated, then we recommend getting a comprehensive genetic screen.
Sweat Test
Sweat chloride testing is the gold standard for the diagnosis of Cystic Fibrosis (Figure 5). The sweat test needs to be done in a CF Foundation accredited center, where personnel is trained to obtain a sweat test. It provides a conclusive CF diagnosis by measuring the concentration of salt in a person's sweat. The simple, painless test is the most reliable way to diagnose Cystic Fibrosis. Infants do not sweat much. Sweat glands have to be stimulated by using an electrical current, along with a chemical called pilocarpine. We usually test on two sides. Often it is both forearms. After the stimulation, a plastic coil device the size of a quarter is placed and covered with gauze to collect the sweat. We advise families to keep babies hydrated and wrapped in a blanket to obtain enough sweat for the test.
After the sweat test is completed, how do you interpret the results? Well, 60 is the magic number. Always think 60 in Cystic Fibrosis. Sweats are measured in milliequivalent per liter units. Anything over 60 is a positive indication of Cystic Fibrosis. Sweat tests between 31 and 59 are intermediate and will need to be repeated. If the repeat sweat is also intermediate, we get a CFTR gene test, either with blood or a buccal swab. Any sweat tests below 30 are consistent with normal findings. As with any test, there is a chance for false positives and false negatives. Some conditions that can cause false positives include hypothyroidism, Addison's disease, and malnutrition. I have seen many malnourished patients with elevated sweats, and once their malnutrition is corrected, their sweat will normalize.
We can also get false negatives if the patient has edema, if the sample was diluted, or if they have milder CF mutations. There are patients with two Cystic Fibrosis mutations of milder severity that can have normal sweat tests. In which case, we follow them annually or every six months to see if they ever develop any signs and symptoms of Cystic Fibrosis.

Figure 5. Sweat Test (Attribution: Prigi Varghese).
Multi-Disciplinary Model
Let's look at the multidisciplinary care model. Multidisciplinary care is when professionals from various disciplines work together to deliver comprehensive care that addresses as many of the patient's needs as possible. As a patient's condition changes over time, the team's composition may change to reflect the changing clinical and psychosocial needs of the patient. This care model was set forth as a standard by the Cystic Fibrosis Foundation. Since Cystic Fibrosis is a complex disease, it requires a team approach. The foundation requires each center to have a core team of nurses, dieticians, physicians, social workers, respiratory therapists, and program coordinators. The rest of the disciplines are recommended but not required. This includes a physical therapist, psychologist, research coordinator, and pharmacist. In keeping with this model, it is not unusual for our team members to go in together to see a patient if they are coming in for a clinic visit.
When I see patients, I go in with the respiratory therapist, the dietician, and sometimes even the GI doctor. Visits go by faster, and patients are appreciative of this team collaboration. This team of various disciplines is also charged with meeting before patient visits to discuss a comprehensive care plan for each patient. At our center, we meet as a team once a week to discuss every patient coming in the next week. This way, each team member knows about any concerns the family has, any abnormal labs, or any changes that will need to occur as part of their care. As part of CF Foundation guidelines, patients over one year of age must have quarterly visits.
Babies are required to have monthly visits until they are six months of age. And then every two months till they turn one. All visits, labs, CF cultures, plans of care are also entered into our center's Cystic Fibrosis registry. This is an extensive registry of CF patients, which includes information on genetic mutations, Clinical features like lung function, CF cultures, height, and weight percentiles, CF medications, respiratory cultures, and any CF-related complications, such as diabetes. The CF registry provides the basis for public reporting of CF Center outcomes, which are provided on the CF Foundation website and are available to families and patients. The registry data is reviewed annually by our whole team, and it helps us to see where we need to make improvements in our care and guides us in our quality improvement projects. The CF Foundation website is www.cff.org.
Mucus, Mucus, Everywhere!
CF affects many parts of the body, not just the lungs. We will look at the central systems affected. This is a great visual of what all CF entails (Figure 6). Sinus problems, salty sweat, trouble breathing, and large hard gallstones, abnormal pancreas function, trouble digesting food, and fatty bowel movements. We will start at the top of the body and work our way down.
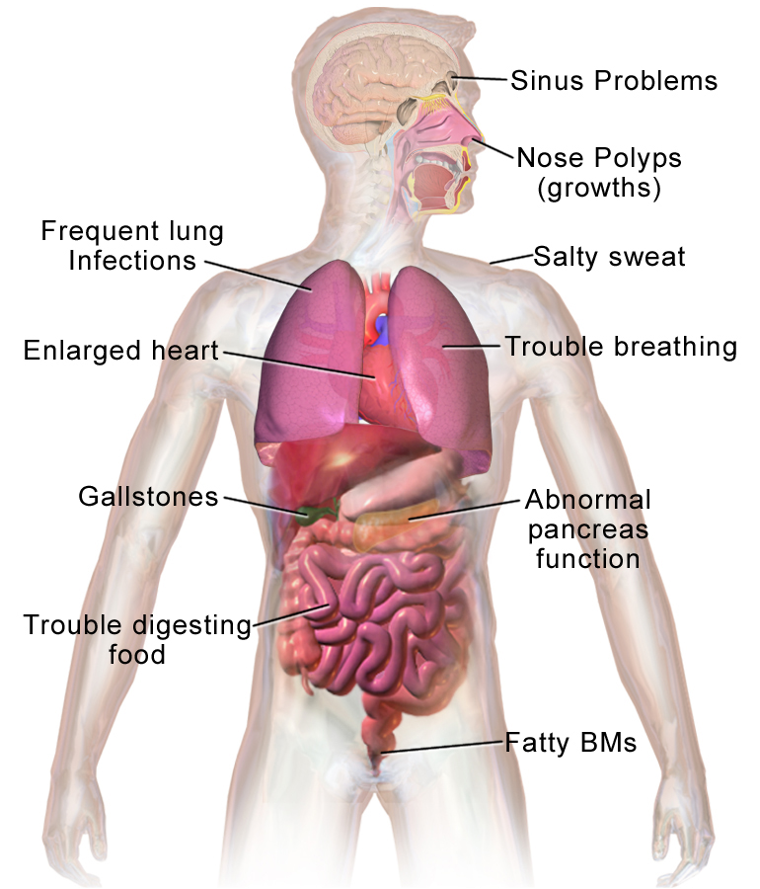
Figure 6. Illustration of body parts affected by CF (Source: Blausen.com staff. Medical gallery of Blausen Medical 2014).
ENT-Sinus Disease
Many CF patients have sinus disease. Normal sinus drainage is when the mucus is thin and efficiently secreted. In CF, the mucus in the sinuses is very thick and sticky. This leads to inflammation and more mucus production (Figure 7). Eventually, the accumulation of bacteria leads to an infection. We treat sinus infections with oral antibiotics, nasal saline sprays, steroids, sinus rinses, and sometimes referrals to the ear, nose, and throat doctors for surgical intervention. Sinuses are often colonized with similar bacteria, as in the lungs. We have had a few older patients be referred to our center by ear ENT doctors because of Pseudomonas in their sinus cavities. That is not normal. When those patients were sweat tested, they were positive for CF.
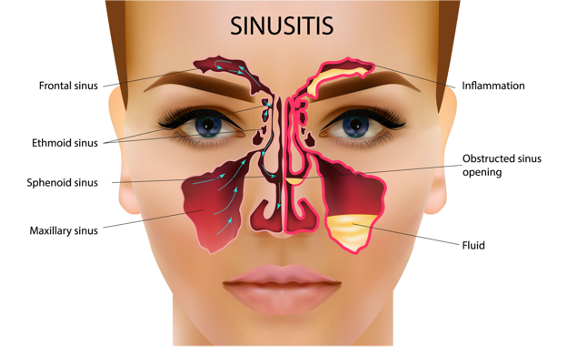
Figure 7. Illustration of sinusitis (Source: Continued licensed from Getty Images).
Nasal Polyps
The other ENT issue that most CF patients have is called nasal polyps (Figure 8). Nasal polyps are soft, painless non-cancerous growth on the lining of the nasal passages or sinuses. They occur due to chronic inflammation due to infection and seasonal allergies. They hang down like teardrops or grapes and are often not bothersome to the patients, but some can be very large and block the nasal passages. It can lead to breathing problems, a loss of sense of smell, and frequent infections. Treatment for nasal polyps is daily use of nasal steroids, sinus rinses, and if bad enough, referral again to the ENT doctor for surgical intervention.
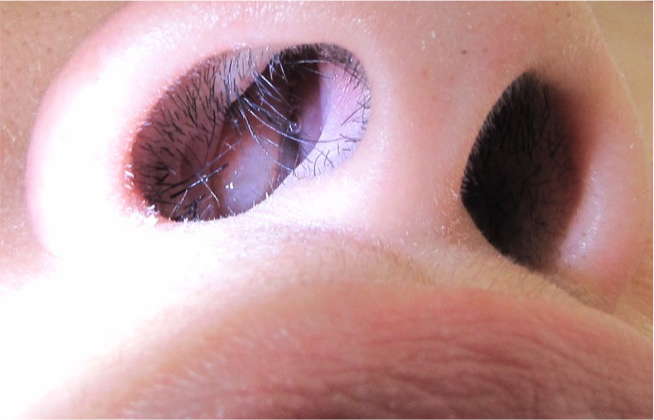
Figure 8. Nasal polyps (Source: MathieuMD via Wikimedia Commons).
NON-CF LUNG
- THIN, RUNNY mucus coats the surface of the airways -produced by the cells lining the breathing tubes
- Tiny hairs called cilia on the surface of the breathing tubes sweep the mucus and foreign particles upward into the larger air passages and then up to the throat where they can be swallowed/coughed up
As this course is geared towards respiratory therapists, the respiratory system is one of the leading systems affected by CF. Mucus is essential in the lungs, where it traps germs and pollutants that we inhale. Cilia, tiny hairs on the outside of the cells, propel the mucus out of the lungs and into the throat, where mucus can be swallowed or coughed out.
CF Lung
- THICK & STICKY mucus makes it hard for cilia to sweep germs & particles out of the lungs leading to persistent lung infections/PNA
- Chronic cough, chronic inflammation leading to airway damage, hemoptysis, Pneumothorax, clubbing, hypoxia, bronchiectasis, mucus plugging
However, in a Cystic Fibrosis lung, the mucus is what? Thick and sticky. And therefore it is very hard for cilia to sweep up the germs and particles from the lungs. As a result, the mucus clogs the airways making it difficult to breathe. Because the mucus in people with CF is also abnormal in other ways, it is less able to kill germs than the mucus in healthy individuals, thus leading to a fertile breeding ground for lung infections, inflammation, and damage. Other symptoms of CF in the respiratory system include hemoptysis, pneumothorax, clubbing of digits, hypoxia, mucus plugging, and bronchiectasis.
I wanted to share a video from the CF Foundation. The video explains what happens when the Cystic Fibrosis transmembrane conductance regulator channel causes this thick, sticky buildup of mucus.
CF Pathogens
Let's look at different CF pathogens or bacteria. Pseudomonas are among the most common bacteria found in people with CF. More than 60% of adults with Cystic Fibrosis have cultured Pseudomonas. It is not a question of if they will get Pseudomonas, but rather when will they get Pseudomonas. When Pseudomonas is established in the airways, it is challenging to eliminate, but aggressive treatment can delay long-term infections. With the first acquisition of Pseudomonas, we do an eradication protocol, including oral Ciprofloxacin for two weeks and inhaled Tobramycin for a whole month.
If patients continue to colonize Pseudomonas, they will be on an antibiotic regimen. Including inhaled Tobramycin every other month or dual therapy, including Tobramycin one month and then another anti-pseudomonal drug, most often Casein, the following month. Other CF pathogens include methicillin-resistant staphylococcus aureus (MRSA), a prevalent bacteria found on skin surfaces. We only treat it during exacerbations. Burkholderia cepacia is another CF pathogen. This germs live in damp places and are often difficult to treat once it infects the lungs. Aspergillus is a common mold fungus found indoors or outdoors. It causes a disease called aspergillosis, which usually only develops in people with weakened immunity or lung disease. Non-tuberculoses mycobacteria (NTM) is another group of bacteria that can affect Cystic Fibrosis patients. This group of bacteria lives in soil, swamps, and water sources. NTM can survive many disinfectants and even severe environmental conditions. These have been found in a growing number of CF people, especially those with advanced lung disease. The treatment for NTM is very lengthy, usually 6 to 12 months with several oral antibiotics, coupled with inhaled antibiotics. Therefore, the CF Foundation guidelines state that all CF patients who can expectorate sputum should have an acid-fast bacilli (ASB) culture sent annually to monitor NTM.
CHEST X-RAY
As RT's, you guys have seen numerous chest x-rays. I wanted to show you how an advanced CF lung can appear on a chest x-ray (Figure 9). You can see the difference, hyperinflation, bronchiectasis, and even areas of consolidation. Results are from the repeated chronic infections that cause weakening of the bronchial walls, leading to bronchiectasis and possible lung collapse.
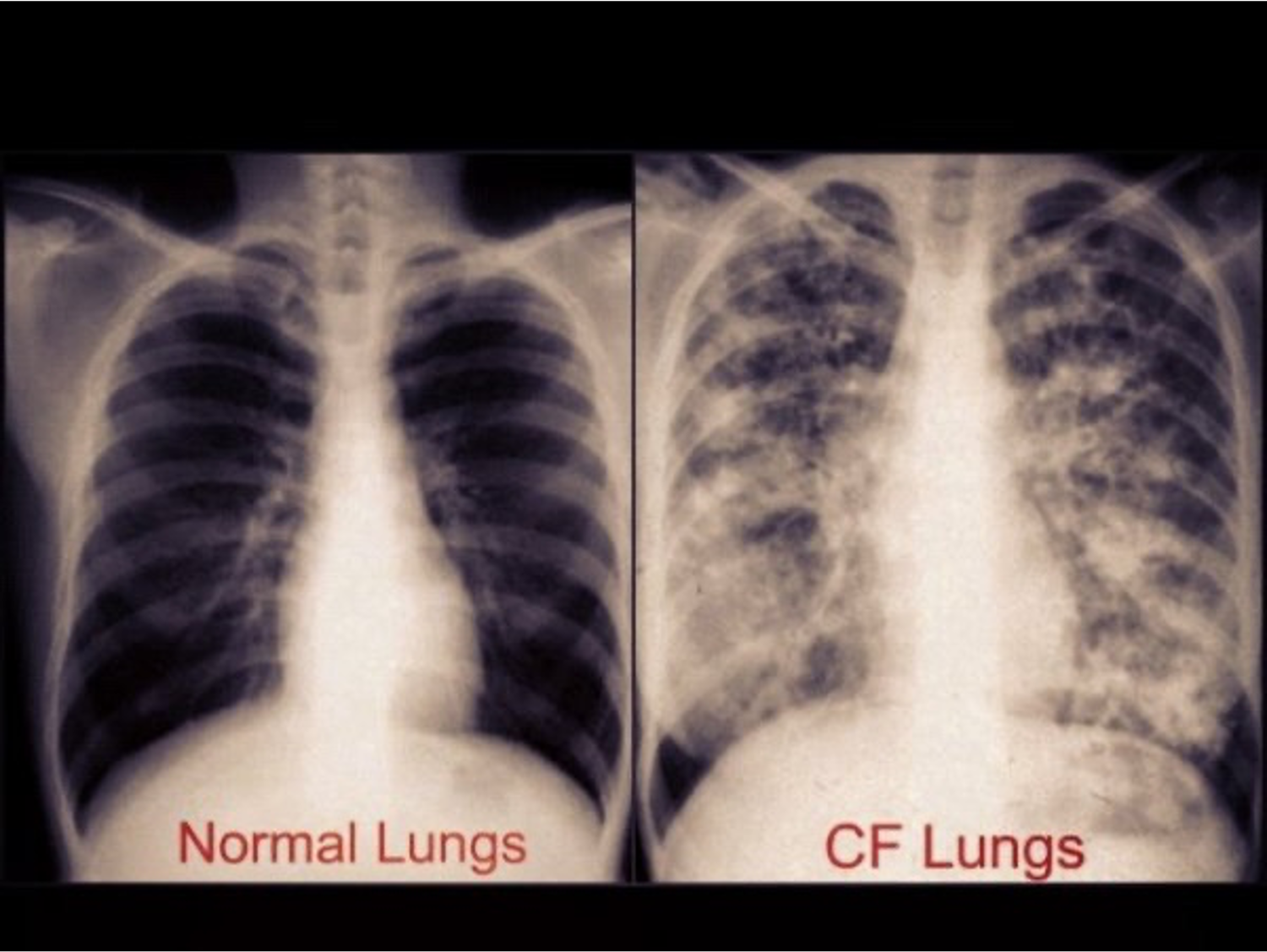
Figure 9. Example of CF chest x-ray.
Pulmonary Exacerbation
- Increased cough
- Increased sputum production
- High fevers ▪ Weight loss> 1 kg
- Increased dyspnea
- Crackles/wheezing
- Decreased exercise tolerance
- Decreased FEV1 on pulmonary function testing
- TREATMENT: outpatient therapy- oral antibiotics x 2 weeks, increased airway clearance, inpatient admission
CF patients are prone to get sicker than those without CF. We call this a pulmonary exacerbation. Our patients come in with an increased cough, increased sputum production, high fevers, weight loss, increased dyspnea, crackles and wheezing, decreased exercise tolerance, and a decreased FEV1 on their pulmonary function tests.
Each patient's exacerbation is different, and therefore we have to tailor therapy accordingly. For example, if there is a mild exacerbation, lung function has not dropped more than 10% from the previous year's best, we might opt for outpatient therapy of increased airway clearance and oral antibiotics. It is usually a two-week course. Then we will see them in the clinic for follow-up. If it is a severe exacerbation and the patient has a significant decline in lung function, weight is down, and the patient may be requiring supplemental oxygen, we will admit, treat with IV antibiotics and aggressive airway clearance. On average, a CF patient's hospital admission is around 10 to 14 days.
Respiratory Management
- Airway clearance: help loosen and get rid of the thick mucus that can build up in the lungs
- Coughing or huffing (a gentle cough)
- Chest physical therapy (CPT), which involves lying in various positions and clapping on the chest Positive expiratory pressure (PEP) device
- Flutter device (also known as oscillating positive expiratory pressure)
- CPT vest
- Exercise
- Bronchodilators
- Mucolytics
- Inhaled Antibiotics
- Steroids
- Aggressive Nutrition
- Monitoring CF sputum cultures/throat swabs
- Pulmonary Function Testing
How do we manage these symptoms? Airway clearance, bronchodilators, mucolytics, inhaled antibiotics, steroids, aggressive nutrition, monitoring CF sputum cultures, and pulmonary function tests are the mainstay for management. We will look at a few of these in detail. First and foremost are airway clearance techniques (ACTs) to clear mucus from the airways.
There are many different ways to clear mucus from the airways, and most are easy to do. Infants and toddlers will need a parent or caregiver's help with their ACTs, mainly because it is one-handed CPT. Older kids and adults can choose airway clearance techniques that they can do on their own. Most of our patients have a CPT vest that vibrates the chest to loosen the mucus. Other options are huff coughing, positive expository pressure devices, such as an acapella or a flutter device, or even aerobic exercise. ACTs are often used along with a bronchodilator. We monitor CF sputum cultures every time they come to the clinic, either for a sick or quarterly visit. When they get admitted, susceptibilities to antibiotics are run on these cultures, and it helps us tailor our antibiotic regimen for each of the CF bacteria they culture.
Vest Therapy
A CPT vest is a machine made up of two pieces an air pulse generator with an inflatable vest connected to the generator by hoses. The generator sends air through the hoses, which causes the best to inflate and deflate rapidly as much as 20 times per second. This rapid inflation and deflation create pressure on the chest, similar to clapping. The vibrations separate mucus from the airway walls and help move it up into the larger airways. Typically, a person uses the vest for five minutes and then coughs or huff coughs to clear the mucus. Sessions can last about 20 to 30 minutes. Patients usually do vest therapy twice a day, more so if they are in exacerbation mode, as they have to be sitting in one place for a lengthy time. Adherence has been an ongoing issue with our patients. However, recently, there have been portable vests on the market, allowing CF patients not to be confined to one area for therapy. It gives some flexibility. These portable vests are for 12 years old and up, as it is a bit heavy. The battery is built into the vest, and therefore we must make sure that our patients have the muscle strength to wear it. We have seen an increase in adherence to vest therapy when patients have switched to these portable vests.
Order of Treatments
- Bronchodilators with CPT vest
- Hypertonic Saline 3% or 7%
- Improves defective mucociliary clearance, increases airway surface liquid, used in patients > 6 years of age
- Pulmozyme: breaks down the DNA & helps reduces the viscosity of mucus in the CF airway
- Inhaled antibiotics
- Steroids
It is safe to say that most CF patients are on a regimen of both bronchodilators and mucolytics. Bronchodilators, Albuterol or Xopenex, should always be given first. Bronchodilators help open up the airways and move air to and from the lungs. Most of our patients both have nebulizers as well as an inhaler. Hypertonic saline is next, and it is extra salty water that is sterile as CF airways are known to lack enough salt and water. The hypertonic saline mist acts as a mucolytic by helping to clear the thick mucus from the lungs. Hypertonic saline is hydrating and increases the airway surface fluid. The 7% solution is used in kids older than six years old. The 3% solution is used in kids under six years old. Some patients do not tolerate hypertonic saline as it is very irritating and sometimes could lead to nosebleeds and even. Others have reported an increase in bronchospasms. Before we start a patient on hypertonic saline in the clinic setting, we usually do a trial. It is recommended to do a hypertonic saline treatment right after the bronchodilator to avoid bronchospasms. The next mucolytic is Pulmozyme (Dornase Alpha). The mucolytic will break down the DNA structure of the thick mucus and thin it out, helping patients cough out secretions that are thinner than their usual mucus. Both these mucolytics are completed twice a day.
After the mucolytics are complete, patients who are on inhaled antibiotics should do those next. Inhaled antibiotics, such as Tobramycin, should be given last. The object of inhaled antibiotics is to attack the bacteria within the lungs. The airways should be open and cleared off as much mucus as possible before receiving this therapy. Some CF patients have an asthma component to their disease, and therefore they are on inhaled corticosteroids. If they are on steroids, this will be the last step in their treatment set. We advise them to rinse their mouth after. Our CF patients have many treatments, and it takes a good 30 minutes to an hour to get at least one set of treatments done. The treatment burden for CF patients is heavy. As providers, we need to be aware and work together with them on a schedule that will work best for them at home.
Pulmonary Function Test
In brief, all PFTs are performed through a tube. The patient takes a big breath in and then breathes it out, explosively as hard and fast as possible, and keeps exhaling until they cannot breathe out any more. The PFT measures lung volume, capacity, and gas exchange. The test repeats at least three times to be sure there is minimal variation. The computer compares the value of these tests done by the person with Cystic Fibrosis with similarly aged persons of the same size and gender without CF and gives us a percent predicted value. The main component of the test we look at is the FEV1, or the forced expiratory volume after one second of blowing out. PFT results interpret as mild, moderate, or severe, and obstructive or restrictive. Most CF patients have obstructive lung disease. In contrast, people with asthma have more of a restrictive process.
We usually start PFT testing at our center at around age five. It takes a while for our patients to get used to the technique and get it down accurately. Recently with the pandemic, the CF Foundation has partnered with a company to send out handheld home spirometers for all of our patients. This way, they can still track and monitor their lung function even at home.
Lung Transplant
Lung transplant is the most aggressive therapy available for end-stage lung disease. The Cystic Fibrosis Foundation guidelines recommend starting the discussion of lung transplant as a future treatment option long before the need ever arises. The guidelines define this as when your patient's forced expiratory volume is in one second, and your FEV1 is less than 50% of predicted. It does not necessarily mean that they need a transplant, only that transplant might be considered an option in the future. An important thing to mention to families and patients is that this is not a cure for CF. Their new lungs will not be affected by CF, but the rest of their organs will continue to be affected. Therefore they will need to continue with other therapies such as their pancreatic enzymes.
Median survival for people with CF who have had a transplant has improved, according to the 2018 International Society for Heart and Lung Transplantation Registry. The median survival for adults has improved from 5.5 years for those transplanted between 1990 and 1998 to 9.5 years for those transplanted between 1999 and 2016. Different factors potentially affect survival in children with CF, for whom the median survival was 5.4 years. For those who received transplants between 1999 and 2016, lung transplantation can extend and improve the quality of life, but it involves an extensive evaluation and dedication to living the lifestyle required to keep the new lungs healthy. The respiratory aspect of CF is enormous, and it requires much preventative care from our patients. As an RT and a provider, we should partner with our patients to help make their care more feasible for home, college, or even work.
How Cystic Fibrosis Affects the Lungs
I wanted to share this video from the CF Foundation on how CF affects the lungs as a wrap-up to the respiratory section of this webinar.
Gastrointestinal System
- Pancreatic Insufficiency ( PI)
- Distal Intestinal Obstructive Syndrome ( DIOS)
- GERD
- Constipation
- Rectal Prolapse
- Meconium Ileus/delayed meconium
- Cystic Fibrosis Liver Disease
As RT's, it is beneficial to know the GI manifestations of CF because you will get a better insight into why your patient may not be doing some of their treatments, why their lung function is still down, or even if they are doing all their treatments. Remember, in Cystic Fibrosis everything is connected.
Pancreatic Insufficiency
The first and probably the most common issue is pancreatic insufficiency. Typically, there is thin mucus around the normal pancreas. However, in Cystic Fibrosis, this mucus is very thick, and it blocks the passage of pancreatic digestive enzymes from reaching the small intestine. Without these enzymes, patients will have malabsorption of fats, proteins, carbs, and fat-soluble vitamins. How can you tell if your patient is pancreatic insufficient? Well, they will have big bulky, oily, foul-smelling, and frequent stools. They will also have abdominal discomfort, pain, and even weight loss.
Pancreatic Enzyme Replacement Therapy (PERT)
How do we fix this? With pancreatic enzyme replacement therapy (PERT), enzymes help digest and absorb fats, proteins, and carbs. Each enzyme capsule contains lipase to break down fats, amylase to break down carbs, and protease to break down proteins. Besides the history of greasy stools, we will send off stool specimens to check for fecal elastase to officially diagnose pancreatic insufficiency. If the fecal elastase is less than 200, the patient is pancreatic insufficient, meaning they need enzymes. If the fecal elastase is greater than 200, they are pancreatic sufficient and do not need enzymes. Some patients with CF mutations that are not as severe or disease-causing will be pancreatic sufficient, and they do not need to be on enzyme therapy.
PERT
As mentioned before, pancreatic enzymes are released into the small intestine. Therefore the enzyme capsules are enteric-coated to withstand stomach acid and will only get activated in the small intestine, which is not as acidic. Patients have to take these enzymes before every meal and every snack. The goal is not to exceed 2,500 units of lipase per kilo per meal.
How Do You Take Enzymes?
Most of our older patients can swallow these capsules, for those who are not, and for our babies, we ask parents to open these capsules and sprinkle the beads into an acidic food, such as apple sauce. If they do not tolerate the apple sauce, we can always use strawberry jam, ketchup, or barbecue sauce. It will preserve the beads from being activated until they reach the small intestine. If patients open and sprinkle the enzymes into their mouth without an agent, it can start to activate and cause erosions and irritation to the mouth and their GI tract.
Side Effects of PERT
As with any medication, there are side effects to enzymes. Allergy to pork is a significant side effect, as all commercially available enzymes are porcine-based. There are research studies right now looking at testing non-porcine-based enzymes for this reason. When we write scripts, patients are informed how many capsules per meal per snack and a maximum to take in a day. A lipase unit of 10,000 per kilo per day is the maximum.
Fibrosing Colonopathy
Taking too many enzymes could lead to something called fibrosing colonopathy—serious side effects of enzymes. Fibrosing colonopathy is associated with severe abdominal pain, diarrhea, and possible rectal bleeding. Diagnosed with a contrast study, it shows radiological hallmarks as a foreshortening of the right colon and varying degrees of stricture formation. Treatment usually is surgical resection of the fibrotic region.
Distal Intestinal Obstruction Syndrome (DIOS)
Another side effect that is just as serious is distal intestinal obstruction syndrome (DIOS). DIOS is when stool accumulates in the colon. There is a palpable right lower quadrant mass with or without pain. An x-ray shows fecal mass in the right iliac fossa and most often mimics appendicitis. With the standardization of enzymes in the 1990s, we see less DIOS. Medical management depends on the severity of the obstruction. Most of the time, treatment is with GoLYTELY clean-out. If that fails, we use the Gastrografin enema, and once the obstruction is cleared, they can go back to the GoLYTELY clean-out. If all of that does not clear the obstruction, the patient will need surgical intervention. Remember, nutrition is a key player in the management of a full-blown CF exacerbation.
High Calorie, High Fat Diet
CF patients use up many calories when they are coughing or fighting an infection. Therefore, all CF patients are on a high-fat, high-calorie diet. They have increased energy needs and fat needs. In other words, they need to eat whatever you and I cannot, such as butter, ice cream, whole milk, pasta, pizza, all of our favorite foods they can have. You would think this would be a fun and easy diet, but most CF patients struggle with weight gain as they do not have the appropriate digestive enzymes. Most of these fat-containing foods will cause abdominal discomfort and pain. When I see patients in the clinic, one of my preferred questions to ask is, what is your favorite food to eat? It is not uncommon for most of them to say salad, tomatoes, or fruits. These are all foods that contain no fat. When they eat this, their stomach does not hurt. As weight gain is a constant issue, we tell parents and patients to increase calories any chance they get. Use coconut oil, double scoops of ice cream, sneak in peanut butter or Ranch into their meals. If that does not work, we have calorie supplements such as Duocal, a powder they can add to their foods to cook with, and Liquigen, which is just pure MCT oil, which adds extra calories to whatever food or drink they like to take.
Oral Supplements
- For Infants
- Standard infant formula and breastmilk have 20 calories per ounce. We can concentrate or fortify both formula and breastmilk up to 30 calories per ounce.
- For Children and Adults
- Scandishakes
- Carnation Instant Breakfast
- Pediasure, Boost Kid Essentials (both 1.0 and 1.5 versions)
- Boost, Ensure (regular and Plus version)
- Resource Breeze, Glucerna
We also use oral supplements, such as Scandishakes or PediaSure Boost, anything to give our patients extra calories. Most often, patients are on oral supplements at least twice a day.
Enteral Supplements
If the patient still has trouble gaining weight or maintaining weight, we recommend placing a gastrostomy tube for overnight feeds. It is a very touchy subject for parents as often they feel they have failed in helping their child gain weight. However, as a team, we are trying to normalize G-tubes as just another tool used in their nutrition care. Nutrition is a critical element in the wellbeing of a CF patient.
Nutritional Impact on FEV1
Average growth is associated with good lung function. The CF Foundation recommends a patient's BMI to be a least in the 50th percentile. Many CF patients struggle with that goal. The higher the BMI, the higher their FEV1. If you look at this graph in Figure 10, the Y-axis is FEV1, and the X-axis is the BMI percentile. We want our patients to be in that high right upper quadrant where their BMI's are high along with their FEV1.
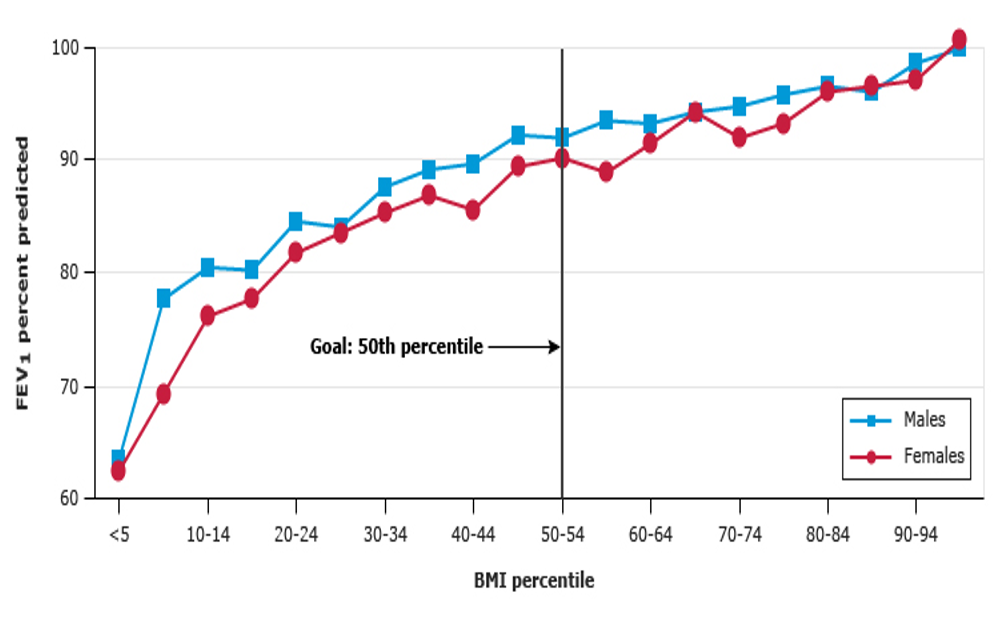
Figure 10. FEV1 versus BMI percentile in children with cystic fibrosis, 6 to 19 years of age (Reproduced with permission from the Cystic Fibrosis Foundation Patient Registry: 2015 Annual Data Report, Bethesda, MD. Copyright © 2016 Cystic Fibrosis Foundation).
ENDOCRINE: Cystic Fibrosis Related Diabetes
As our CF patients are on high fat, high-calorie diets, we need to monitor CF-related diabetes. We know that in type I diabetes body is not making any insulin. In type II, the body cannot use insulin properly and often needs more. Cystic Fibrosis related diabetes shares some features with both type I and type II diabetes. In people with Cystic Fibrosis, the thick, sticky mucus that is characteristic of the disease causes scarring of the pancreas. This scarring prevents the pancreas from producing normal amounts of insulin. So like people with type I diabetes, they become insulin deficient. Their pancreas still makes some insulin, but not enough to stay healthy and maintain good nutrition.
Diagnosis of CFRD
How do we screen and diagnose CF-related diabetes? The CF Foundation recommends an annual oral glucose tolerance test, starting at age 10. Diagnostic for CFRD when they are oral glucose tolerance test two hour glucose is above 200. If they fit diagnostic criteria, we refer our patients to our endocrinologist. If they are impaired, we teach them how to monitor blood sugars a couple of times a week. Then we will repeat the oral glucose tolerance test. Management for Cystic Fibrosis related diabetes is straight insulin. There is no restriction to their food, and they do not go on a diet. They continue on their high fat, high-calorie diet.
Bone Health
Going hand in hand with nutrition is exercise and bone health. Vitamin labs are checked annually and as needed. It is not uncommon for our patients to be on extra vitamin D supplements or calcium supplements. The CF Foundation recommends DEXA scans starting at age eight and then completed annually. The DEXA scan or bone density is a short, painless task lasting about 30 seconds per scan. The scan assesses the bone mineral density of different types of bone in a person's body. The scan also provides a measure of fracture risk. As mentioned before, exercise is also an excellent substitute for the best therapy. In our center, we have a physical therapist who will see patients who meet the criteria for low bone density and those with advanced lung disease. Most of our patients see physical therapy, outpatient, at least twice a week.
Reproductive System
Another system affected is the reproductive system. Male infertility is a significant issue with Cystic Fibrosis. About 96% of men are sterile. It is not that they do not make sperm. They lack the vas deferens that carry the sperm. Puberty is delayed in both males and females, mainly related to nutrition. As in other body systems, mucus in the cervix is also very thick and abnormal, making pregnancy harder. Though, I have seen adult CF patients who have gone on to have successful pregnancies. Otherwise, patients with Cystic Fibrosis go the route of IVF to have kids.
Psychosocial Issues
CF also takes a toll on mental health. Most of these patients spend long periods in the hospital with their admissions. They also have to set apart hours in a day to do treatments. Frequently, they are isolated from others, especially those CF patients that they can connect with. It all leads to increased anxiety, depression, and poor adherence to therapies. New CF Foundation guidelines for mental health require annual anxiety and depression screening for patients and caregivers at 12 years of age. In our center, we have a psychologist who can see all of our patients that need mental health support.
Infection Control
Infection control has been a hot topic for our CF patients even before the pandemic. Many germs are hazardous for people with Cystic Fibrosis, and they lead to a faster decline in lung function. Medical studies have shown that people with Cystic Fibrosis are at particular risk of spreading certain germs among others who have the same disease, called cross-infection. Most important is that all CF patients are on contact precautions. They must wear a mask when walking in the halls of the hospitals or the waiting rooms. They do not have to wear a mask when they are in their rooms. Hospital staff must wear a gown, gloves and now wear a mask when seeing CF patients. Rooms and stethoscopes are cleaned in between patients. Of course, hand hygiene must be performed before and after patient contact. The six feet rule only applies to the patients. Experts chose the distance of six feet because this is how far germs can spread. When a person coughs, sneezes, or even speaks, keeping six feet away from someone else who is sick helps keep a Cystic Fibrosis patient from catching whatever bacteria or virus they may have.
Although the infection control guidelines can add to feelings of isolation and make it even more difficult for people with CF to connect. I am happy to say that many CF centers have developed strategies such as online social networking, support groups via Skype or FaceTime, and even CF blogs to help patients connect. In addition, the CF Foundation has programs like CFPR Connect and virtual events that provide space for people with CF and their family members to build relationships in real-time with those who understand their experiences the best.
New Drugs for CF
The medications we have reviewed thus far, the mucolytics, the enzymes, the vitamins are all meds that treat the symptoms of Cystic Fibrosis. Now let's look at a new drug that is on the market for CF. Cystic Fibrosis transmembrane conductance regulator modulator therapy is designed to correct the malfunctioning protein made by the CFTR gene. Because different mutations cause different defects in the protein, as we learned earlier, the medications that have been developed so far are only effective in patients with specific CF mutations. With the increase in research, 80% of all CF patients are eligible for a modulator. These modulators work on the cell surface to correct the defect and halt the progression of Cystic Fibrosis. The first modulator Kalydeco came out in 2012 and was a game-changer. The latest modulator is a triple combination drug called Trikafta, which came out first in October of 2019, and it was only for patients 12 years and older. In June of 2021, Trikafta got approved for six years and older. It has changed the lives of CF patients everywhere. Trikafta quickly increases on average lung function by about 10 to 15%. I have personally seen patients get taken off transplant referral lists due to this drug. It is as close to a cure for CF as we can get right now. Yet, we do have 10% of the population that is not eligible for a modulator. The CF Foundation continues to press on with research until the modulator is available for all patients with Cystic Fibrosis. During the latter part of adolescence, most people living with Cystic Fibrosis transfer from a CF care center specializing in treating children to one specializing in treating adults with Cystic Fibrosis.
Transition
Most people would see a transition from pediatric to adult care between the ages of 17 and 21. We usually transition to the adult center in our center once our patients have completed the first semester of college. We work closely with our adult team to make sure that the transition is smooth as possible. At our center, we are working on having our adult patients come to the clinic by themselves and start taking ownership of their health and care management. On their last visit to our center, they receive a book, "Oh, The Places You Will Go," signed by the entire care team. It is wonderful to see our patients transition to the adult center.
CF Survival
Between the 1980s and 1990s, the life expectancy of people with Cystic Fibrosis was in the teens 10 to 14 years of age. As you can see on this graph in Figure 11, with each new drug and therapy developed, the life expectancy increases. Based on 2019 registry data, the life expectancy of people with Cystic Fibrosis born between 2015 and 2019 is 46 years. Data also shows that babies born in 2019 with CF are predicted to live to be at least 48 years or even older. These numbers give hope to those with CF and caregivers because of tremendous advancement in research and care. Many people with CF live long enough to realize their dreams of attending college, pursuing careers, getting married, and having kids. While there has been significant progress in treating this disease, there is still no cure for CF, and 46 years or 48 years is not good enough. The CF Foundation will continue to conduct many clinical trials and invest in companies working on modulator therapy and gene therapy so that one day CF will stand for cure found.
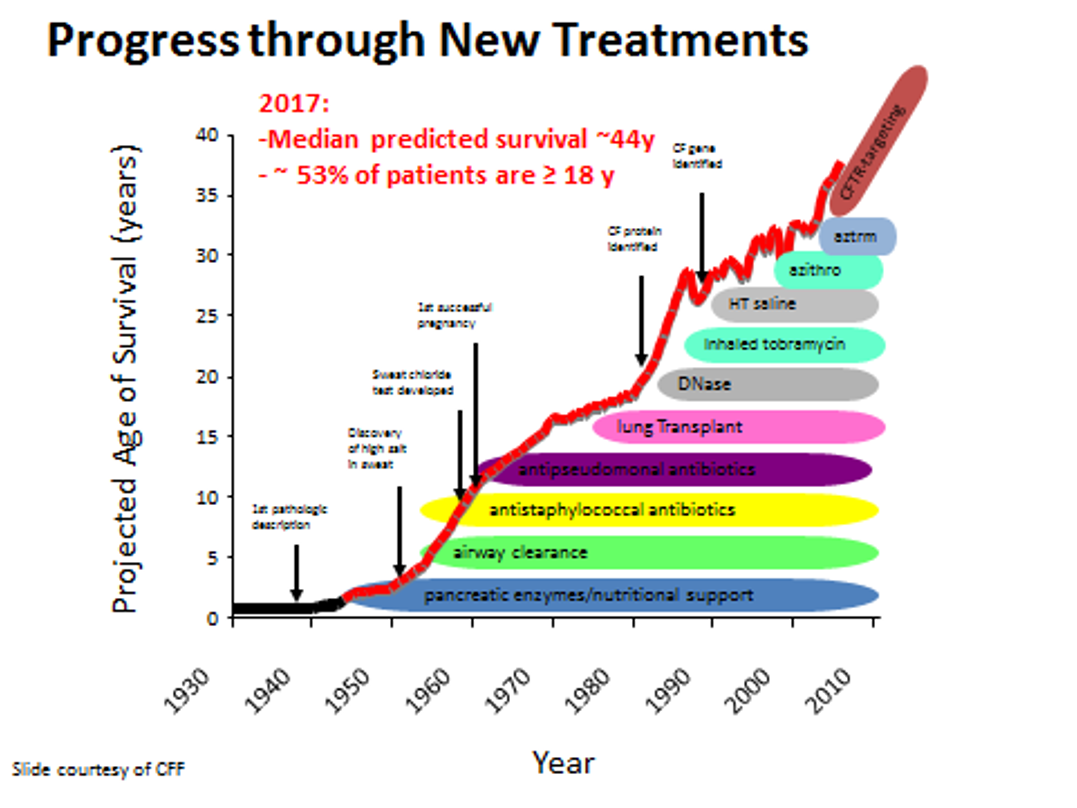
Figure 11. Graph of projected age of survival (Source: Image courtesy of Cystic Fibrosis Foundation).
Resources
References
Borowitz et al. Gastrointestinal outcomes and confounders in Cystic Fibrosis. JPGN 2005 Sept, 41: 273-285.
Borowitz, D. Evidence for the diagnosis of pancreatic insufficiency. Pediatric Pulm, 29: 167-168.
Bryson, E., Feenan, L. (2014). Nuts & Bolts of CF Care (28th Annual North American Cystic Fibrosis Conference). Atlanta, GA.
Cunningham, J., & Taussig, L. (2013). An Introduction to Cystic Fibrosis: For Patients and their Families (6th ed.). Bethesda, MD: Cystic Fibrosis Foundation.
Orenstein, D. (2004). Cystic Fibrosis: A Guide for Patient and Family (3rd ed.). Philadelphia, PA: Lippincott Williams & Wilkins.
Rho J, Ahn C, Gao A, Sawicki GS, Keller A, Jain R. Disparities in Mortality of Hispanic Patients with Cystic Fibrosis in the United States. A National and Regional Cohort Study. Am J Respir Crit Care Med. 2018 Oct 15;198(8):1055-1063.
Citation
Varghese, P. (2021). Cystic fibrosis: a comprehensive review. Continued.com - Respiratory Therapy, Article 112. Available at www.continued.com/respiratory-therapy
