Editor’s note: This text-based course is an edited transcript of the webinar, SMA in Pediatrics (Spinal Muscular Atrophy), presented by Nadia Boussalham, BS, RRT.
It is recommended that you download the course handout to supplement this text format.
Learning Outcomes
After this course, participants will be able to:
- Recognize Spinal Muscular Atrophy (SMA) and review its implications in pediatric patients
- Discuss the different types of Spinal Muscular Atrophy (SMA) and identify the available medications for treatment
- Describe respiratory treatments and management strategies for pediatric patients diagnosed with Spinal Muscular Atrophy (SMA)
What Happens With SMA?
To begin with, Spinal Muscular Atrophy (SMA) is a genetic disorder that causes muscle weakness, particularly in the skeletal muscles. As a result, it can severely affect a child's ability to crawl, walk, or even control head movements. In more severe cases, SMA can impair the muscles involved in breathing and swallowing, posing serious health risks.
The root of the problem lies in the motor neurons, which control muscle strength and movement. These neurons are located in the spinal cord and the lower part of the brain. In individuals with SMA, these motor neurons start to break down, preventing the brain’s signals from reaching the muscles. As a result, the muscles weaken and eventually deteriorate, a process known as atrophy, similar to what occurs in other genetic disorders.
SMA is an autosomal recessive disorder, meaning a person must inherit two mutated genes—one from each parent—to develop the condition. Most forms of SMA are linked to a mutation in the SMN1 gene, which fails to produce sufficient protein to support the motor neurons. Without enough of this protein, the neurons degenerate and lose their ability to send signals to the muscles, leading to progressive muscle weakening and damage.
Genetic Disease
A child with spinal muscular atrophy (SMA) inherits one copy of the mutated gene from each parent. For a child to develop SMA, they must inherit two non-functioning SMN1 genes—one from each parent. This pattern of inheritance is known as autosomal recessive, meaning both copies of the gene must be defective for the disease to manifest.
If both parents are carriers of the same SMN1 gene mutation, there is a 25% (or one in four) chance that the child will inherit the mutated gene from both parents, resulting in the child developing SMA. This is because each parent has one functioning and one non-functioning gene copy, and the likelihood of both non-functioning genes being passed down simultaneously is 25%. This scenario can happen with no prior symptoms in the family, as carriers of the mutation are asymptomatic.
In addition to the 25% chance of having a child with SMA, there is a 50% chance that the child will inherit only one copy of the mutated gene, making them a carrier, like their parents. Carriers of the SMA gene do not exhibit symptoms of the disease but can pass the mutated gene to their children. This means that while carriers themselves are unaffected, they still have a 50% chance of passing the defective gene to the next generation, increasing the likelihood of SMA in future descendants if both parents are carriers.
Lastly, there is a 25% chance that the child will inherit two fully functioning SMN1 genes, one from each parent, meaning the child will neither have the disease nor be a carrier of the mutated gene. In this case, the child is entirely free from the SMA gene mutation and its associated risks.
It is crucial to understand the value of carrier testing, which can identify individuals who carry the SMN1 gene mutation. Carrier testing is particularly important for families with a history of SMA or for couples planning to have children. Early knowledge of carrier status allows for genetic counseling, where healthcare professionals can help prospective parents understand the risks and implications of SMA, guide them in making informed reproductive decisions, and discuss potential options like in vitro fertilization (IVF) with pre-implantation genetic diagnosis (PGD) to select embryos without the mutation.
Additionally, prenatal testing can be conducted during pregnancy to determine if the fetus has inherited the mutated genes. This can provide critical information for managing pregnancy and preparing for potential outcomes if the child is diagnosed with SMA.
Overall, understanding the inheritance pattern of SMA and the options available for carrier testing and genetic counseling can help families make proactive choices and manage risks associated with this genetic disorder.
Severe Neuromuscular Disease
SMA is a severe and progressive neuromuscular disease that primarily affects motor neurons, leading to muscle weakness and atrophy. According to the Centers for Disease Control and Prevention (CDC), as of 2021, approximately one in every 10,000 births result in a baby being born with SMA, making it one of the most common causes of genetic infant mortality. Furthermore, an estimated one in 51 people worldwide is a carrier of the SMA gene mutation, underscoring its prevalence as one of the most common rare diseases.
This frequency places SMA alongside other well-known genetic conditions such as cystic fibrosis, Duchenne muscular dystrophy, and amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease. Like these conditions, SMA can have a profound impact on motor function, with the severity of symptoms varying based on the specific type of SMA and the number of copies of the SMN2 gene a person carries.
SMA affects individuals across all genders, races, and ethnicities, with no disparities in its prevalence. Both males and females are equally likely to inherit the disease, as SMA is passed down through an autosomal recessive inheritance pattern. This means that for a child to develop SMA, they must inherit two copies of the defective SMN1 gene—one from each parent. If both parents are carriers of the mutation, there is a 25% chance that their child will inherit both faulty genes and develop SMA, a 50% chance that the child will be a carrier like the parents and a 25% chance that the child will inherit two normal copies of the SMN1 gene.
The diagnosis of SMA is confirmed through molecular genetic testing, a highly reliable method that identifies mutations in the SMN1 gene responsible for the disorder. Genetic testing typically involves a blood sample, and in some cases, prenatal genetic testing may be offered if there is a family history of SMA or if both parents are known carriers. In recent years, many states have also included SMA in their newborn screening programs, allowing for early detection before symptoms appear. Early diagnosis is critical because it enables early intervention with treatments that can significantly improve outcomes, particularly with the advent of gene therapy and splice-modifying therapies.
Diagnosis of SMA
Testing for mutations in the SMN1 and SMN2 genes is highly reliable and considered the gold standard for diagnosing spinal muscular atrophy (SMA) when typical symptoms are present. Because of the accuracy of genetic testing, there is usually no need for a muscle biopsy. Genetic testing involves a quantitative analysis of both SMN1 and SMN2 genes using a technique known as multiplex ligation-dependent probe amplification (MLPA). This method allows for the detection of copy number variations in both genes.
Another commonly used method is PCR followed by restriction digest, which can identify homozygous deletions in the SMN1 gene. While PCR is faster and less expensive than MLPA, it does not provide a quantitative analysis of SMN1 or SMN2 copy numbers. However, it is often readily available in many labs and remains a reliable diagnostic option.
Understanding the number of SMN1 copies is crucial for identifying heterozygous deletions, while the number of SMN2 copies plays a key role in predicting disease severity and guiding therapeutic approaches. In diagnosing SMA, it is important to note that some of its symptoms can resemble those of other neuromuscular disorders, such as muscular dystrophy. To determine the cause of these symptoms, healthcare providers will typically conduct a physical exam and obtain a comprehensive medical history. Physicians may also order additional tests, including genetic testing, to confirm the diagnosis of SMA.
Some of the tests used to diagnose SMA include blood tests, such as enzyme and protein tests, which check for elevated levels of creatine kinase. This enzyme is released into the bloodstream when muscles deteriorate, serving as a potential indicator of muscle damage. A genetic test is another critical diagnostic tool specifically designed to identify mutations in the SMN1 gene. Genetic testing is highly effective, with a 95% success rate in detecting altered SMN1 genes.
In some states, SMA is included in routine newborn screening, which may also involve nerve conduction tests, such as an electromyogram (EMG). An EMG measures the electrical activity of nerves and muscles, helping to assess neuromuscular function.
In rare cases, a muscle biopsy may be performed. This procedure involves removing a small sample of muscle tissue and sending it to a lab for analysis. The biopsy can reveal muscle atrophy, which, as previously mentioned, is the progressive loss of muscle tissue.
But why does this happen? As we have discussed, spinal muscular atrophy is a genetic disorder. Most forms of SMA are caused by mutations in the survival motor neuron (SMN) gene, which leads to the degeneration of motor neurons and the resulting muscle weakness and atrophy.
Why Does It Happen?
The SMN1 gene, located on the fifth chromosome, is crucial for producing the survival motor neuron (SMN) protein, which plays an essential role in the health and function of motor neurons. These motor neurons are responsible for transmitting signals from the brain to the muscles, allowing for voluntary muscle movements. When the SMN1 gene is functioning normally, it produces sufficient levels of the SMN protein to maintain normal motor neuron function, enabling muscles to receive the necessary signals to move.
However, when the SMN1 gene is mutated or deleted, there is an insufficient production of SMN protein, leading to the degeneration of motor neurons. This deterioration disrupts the brain's ability to communicate with the muscles, ultimately causing the characteristic muscle weakness and wasting (atrophy) seen in SMA. The progressive loss of motor neurons results in the muscles' inability to contract and move, affecting a person's mobility and, in severe cases, their ability to breathe and swallow.
The severity of SMA is heavily influenced by the number of copies of the SMN2 gene, a gene that is similar to SMN1 but produces less functional SMN protein. The SMN2 gene is considered a "backup" gene because, while it cannot fully replace SMN1, it can still produce a small amount of functional SMN protein. The number of SMN2 copies a person has varies from individual to individual, and this variation significantly impacts the severity and progression of the disease.
Individuals with fewer copies of SMN2 (typically one or two copies) tend to experience the most severe forms of SMA, such as Type 1 SMA, which manifests in infancy. These individuals produce very little functional SMN protein, leading to the early onset of symptoms, including severe muscle weakness, difficulty breathing, and problems with swallowing.
Individuals with more copies of SMN2 (three or more copies) generally have a later onset and a milder disease course, as seen in Type 2 or Type 3 SMA. These individuals produce more SMN protein, which provides some level of compensation for the loss of SMN1 function. This results in less severe symptoms, such as delayed motor milestones (e.g., sitting, standing, walking), but they still experience progressive muscle weakness over time.
In Type 4 SMA, which typically occurs in adulthood, individuals may have up to six or eight copies of SMN2, allowing for a relatively mild progression of the disease with less impairment in daily activities compared to earlier-onset forms.
While the SMN2 gene provides some compensation for the lack of SMN1, most of the SMN protein it produces is truncated and non-functional due to a splicing error that removes exon 7. Only a small fraction of the protein produced by SMN2 is full-length and functional. Therefore, the more copies of SMN2 an individual has, the more functional SMN protein is produced, which helps mitigate the severity of the disease but does not fully prevent the symptoms of SMA.
What Is A Gene?
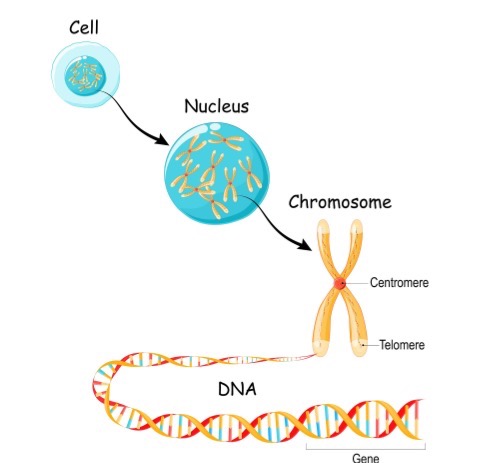
Figure 1. Illustration of a gene (Continued, Getty).
A gene is the basic physical and functional unit of heredity, made up of DNA (Figure 1). Some genes provide instructions for making molecules called proteins, which are essential for the body’s structure and function. In humans, genes vary widely in size, ranging from a few hundred DNA bases to over 2 million bases.
Humans have approximately 20,000 to 25,000 genes—that is a lot of genetic material! To keep track of them, scientists give each gene a unique name. Since gene names can be long, they are often assigned symbols, which are short combinations of letters and sometimes numbers that serve as an abbreviated version of the full gene name. For example, a gene on chromosome seven associated with cystic fibrosis is called the cystic fibrosis transmembrane conductance regulator, but its symbol is CFTR.
Gene Defect in SMA
- Patients with SMA lack a functioning SMN1 gene
- The SMN1 gene tells the body how to make survival motor neuron (SMN) protein
Patients with SMA lack a functioning SMN1 gene. The SMN1 gene provides instructions for the body to produce SMN protein, which is essential for maintaining the health and function of motor neurons.
To give an example, most people are familiar with cystic fibrosis, a genetic condition linked to a specific gene mutation. Similarly, the most common form of SMA is caused by a mutated or missing SMN1 gene. This gene is located on chromosome five and produces the SMN protein, which is critical for motor neuron survival. When a mutation occurs in both copies of the SMN1 gene (one on each chromosome five), there is a deficiency in SMN protein production. Normally, most proteins produced by SMN1 are full-length and functional, but in SMA, little to no full-length, functional SMN protein is made.
However, this loss can be partially compensated by the presence of neighboring SMN2 genes, which are 99% similar to SMN1. The number of SMN2 gene copies varies between individuals. Although most proteins produced by SMN2 are short and non-functional, some are full-length and functional. The more copies of SMN2 a person has, the more functional SMN protein is produced, which can moderate the severity of the disease.
SMA is a neuromuscular disorder characterized by this deficiency in SMN protein, leading to the degeneration of motor neurons and progressive muscle weakness. Understanding the role of both SMN1 and SMN2 is essential for grasping the genetic basis and variability of the disease.
SMN Protein
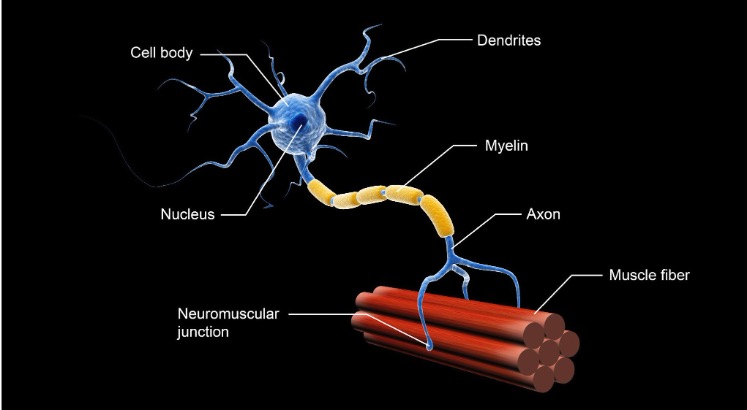
Figure 2. Structure of a motor neuron (Continued, Getty).
Figure 2 illustrates the structure of a motor neuron. The motor neuron dysfunction in SMA leads to progressive muscle weakness and atrophy, with the severity typically greater in the proximal muscles—those closer to the center of the body, such as the muscles in the trunk, chest, upper legs, and arms. In contrast, the distal muscles, such as those in the hands and feet, are generally less affected. This pattern of muscle weakness makes tasks like standing, walking, or lifting objects more difficult, especially as the disease progresses.
The SMN protein, which is deficient in SMA, plays a critical role in processing molecules called messenger RNA (mRNA). mRNA is essential in the process of protein synthesis, where genetic information from DNA is translated into functional proteins. The SMN protein is specifically involved in the assembly of small nuclear ribonucleoproteins (snRNPs), which are necessary for the accurate splicing of pre-mRNA. Proper splicing ensures that mRNA is correctly formed, allowing the production of functional proteins. Additionally, the SMN protein supports the development and maintenance of dendrites and axons, which are extensions of nerve cells (neurons) that facilitate the transmission of signals between the brain, spinal cord, and muscles.
For example, the phrenic nerve, which originates in the neck and extends to the diaphragm, sends involuntary signals to the diaphragm to control breathing. In severe forms of SMA, the degeneration of motor neurons that control the phrenic nerve can compromise respiratory function, making it difficult for affected individuals to breathe independently. This is why many patients with severe SMA, particularly Type 1, often require respiratory support, such as non-invasive ventilation or mechanical ventilation, to assist with breathing.
The SMN2 gene is considered the disease-modifying gene in SMA because it is almost identical to SMN1, except for a single nucleotide difference in exon 7. This small difference has significant consequences for the processing of SMN2 mRNA. In most cases, the exclusion of exon 7 during the splicing process results in the production of truncated, non-functional versions of the SMN protein. However, SMN2 can still produce some functional SMN protein, as one of the versions, known as isoform D, includes exon 7 and is full-length and functional.
Isoform D accounts for approximately 10% of the total functional SMN protein produced by SMN2. While this small amount of protein can help mitigate the severity of SMA, it is insufficient to fully compensate for the loss of SMN1. The more copies of SMN2 a person has, the greater the amount of isoform D that can be produced, which helps increase the overall levels of functional SMN protein in the body. This is why individuals with more copies of SMN2 tend to have a milder form of SMA with a later onset of symptoms, as their bodies can produce more functional SMN protein.
In severe cases of SMA, such as Type 1, patients may have only one or two copies of SMN2, leading to very low levels of functional SMN protein. This results in early-onset symptoms, such as profound muscle weakness, difficulty swallowing, and respiratory complications. On the other hand, patients with Type 3 or Type 4 SMA often have three or more copies of SMN2, allowing for a higher production of isoform D and thus a milder disease course with later onset symptoms and slower progression of muscle weakness.
SMN2 Gene
The SMN2 gene produces several different versions of the SMN protein, most of which are shorter and quickly degraded due to the exclusion of exon 7 during the splicing process. These truncated proteins are non-functional and cannot adequately support motor neuron survival. Only a small fraction of the protein produced by SMN2 is full-length and functional, which contributes to the overall amount of functional SMN protein in the body.
The number of SMN2 gene copies a person has can vary between individuals, ranging from two to eight copies, and this number plays a crucial role in determining the severity and type of SMA a person develops. The more copies of SMN2 a person has, the more functional SMN protein they can produce, which helps mitigate the severity of the disease.
- Type 1 SMA (the most severe form of the disease) is typically associated with two copies of the SMN2 gene. Patients with Type 1 SMA usually show symptoms early in infancy, such as severe muscle weakness, respiratory issues, and difficulty swallowing. These patients often require respiratory support and have a limited ability to achieve motor milestones, such as sitting or standing without assistance.
- Type 2 SMA patients usually have three copies of SMN2. This form of the disease presents in early childhood, with symptoms appearing between six months and two years of age. While Type 2 patients may be able to sit independently, they generally cannot walk unaided and will experience progressive muscle weakness over time.
- Type 3 SMA, which can manifest later in childhood or adolescence, is also associated with three copies of SMN2 but tends to be less severe than Type 2. Within Type 3, there is a subcategory known as Type 3b SMA, which is linked to four copies of SMN2. Individuals with Type 3b typically experience a later onset of symptoms and may retain the ability to walk longer than those with fewer SMN2 copies, though they will still experience progressive muscle weakness.
- Type 4 SMA, the mildest form of the disease, usually appears in adulthood and is linked to four to six copies of SMN2. Patients with Type 4 SMA may retain most of their motor function for a significant portion of their lives and tend to experience slower progression of muscle weakness compared to those with earlier-onset forms of the disease.
The relationship between SMN2 copy number and the severity of SMA is a key factor in understanding the variability of the disease. In patients with more SMN2 copies, the increased production of full-length SMN protein provides greater support for motor neuron function, leading to a milder disease course. However, despite the presence of additional SMN2 copies, none of the forms of SMA can be completely prevented, as SMN2 cannot fully replace the function of the SMN1 gene.
As we continue, we will delve deeper into the different types of SMA and discuss their clinical characteristics, treatment strategies, and management approaches to help better understand the variability and progression of this complex neuromuscular disorder.
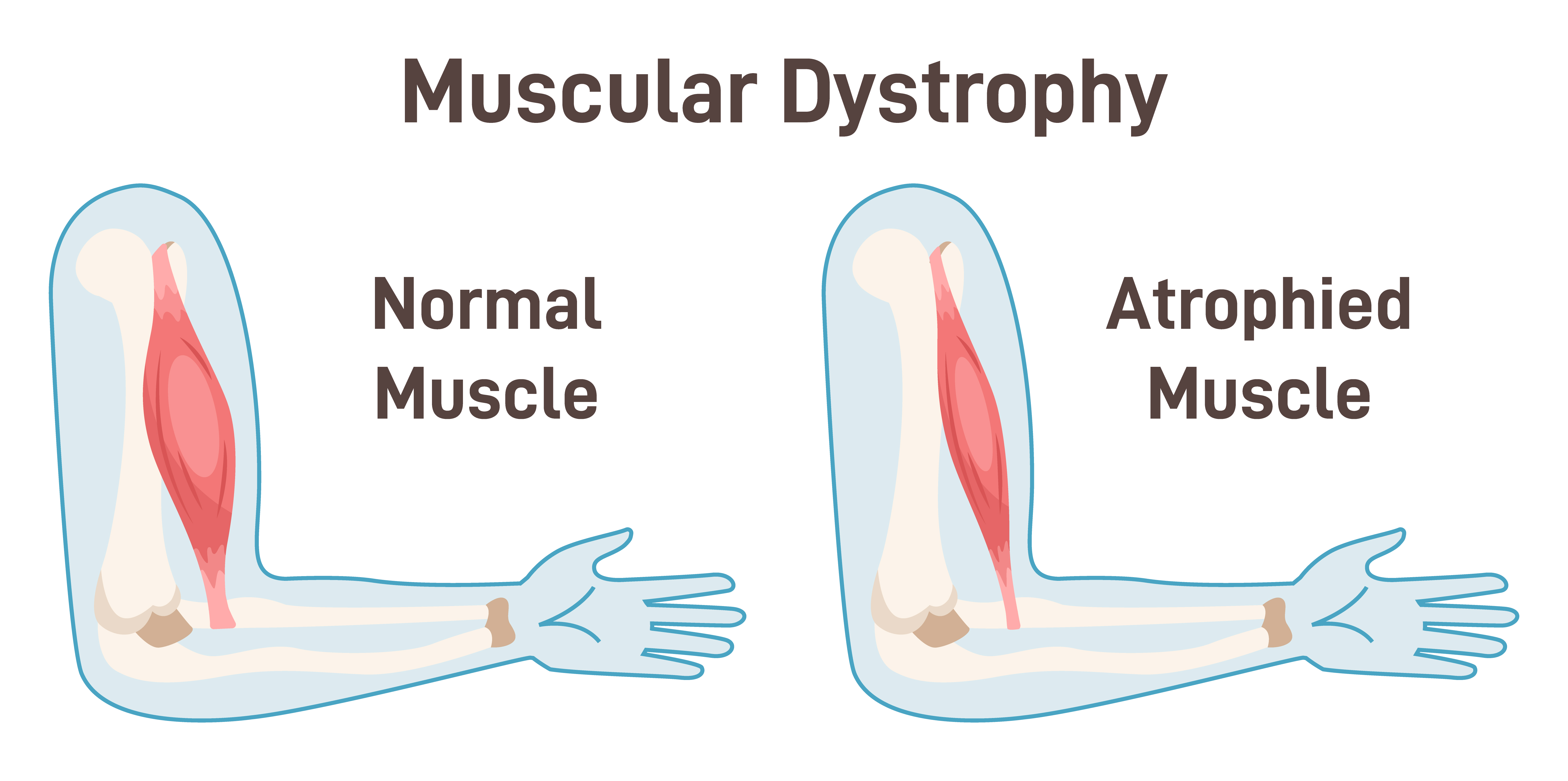
Figure 3. Illustration of normal muscle and atrophied muscle (Continued, Getty).
In SMA, the deterioration of motor neurons—the nerves responsible for controlling muscle strength and movement—leads to progressive muscle weakness (Figure 3). These motor neurons are located in the spinal cord and the lower part of the brain. As they break down, they lose the ability to transmit signals from the brain to the muscles. Without these signals, muscles are unable to contract, leading to a gradual loss of muscle mass, a process known as atrophy.
Muscle weakness in SMA is usually symmetrical, meaning it affects both sides of the body equally. The weakness tends to be more severe in the proximal muscles, which are the muscles closest to the body’s core, such as those in the shoulders, hips, thighs, and upper arms. In contrast, the distal muscles, such as those in the hands and feet, are often less affected. This pattern of weakness makes it difficult for individuals with SMA to perform tasks like lifting objects, walking, or even holding up their heads in severe cases.
As the muscles atrophy, they become visibly smaller and thinner. In advanced stages of SMA, this muscle wasting can lead to significant physical impairments, including difficulty with basic motor functions, such as sitting up or moving the limbs. Over time, the lack of muscle activity can also result in complications such as joint contractures (stiffening of joints due to shortened muscles) and scoliosis (curvature of the spine), which can further limit mobility.
In addition to muscle weakness, the breakdown of motor neurons in severe cases of SMA can also affect respiratory muscles, particularly the diaphragm, making it difficult for patients to breathe independently. This is why many patients with more severe forms of SMA may require respiratory support, such as non-invasive ventilation or mechanical ventilation, to assist with breathing. As muscle atrophy in SMA is a gradual process, early intervention through physical therapy, assistive devices, and modern treatments can help slow the progression and improve the patient’s quality of life.
Staying Ahead of the Progression of SMA Today
SMA is often discovered through newborn screening, which has become a routine part of early diagnostics in many countries. If a child tests positive for SMA during this screening, doctors will typically order genetic tests to confirm the diagnosis and determine the number of copies of the SMN2 backup gene that the child has. These tests may also detect specific genetic mutations that indicate whether the SMN1 gene is malfunctioning or missing entirely. The number of SMN2 copies is crucial because it plays a significant role in predicting the severity of SMA—the more copies a person has, the milder the progression of the disease is likely to be. Identifying the number of SMN2 copies before a baby shows any symptoms is vital for initiating early treatment, which can significantly alter the course of the disease.
For example, if left untreated, a baby with only two copies of SMN2 is likely to show symptoms much earlier than a baby with four copies. In the past, before newborn screening was widely implemented, most children weren’t diagnosed with SMA until they began showing symptoms. At that point, the child was assigned a "type" of SMA based on when symptoms first appeared and which motor milestones they had or had not reached. Since newborn screening wasn’t always routine, this classification was largely based on observation and, in many cases, a degree of guesswork. The earlier the onset of symptoms, the more severe the type of SMA was assumed to be. There are currently four main types of SMA, with an additional fifth type that can be classified as Type 0 or congenital SMA.
By screening newborns and identifying SMA before symptoms appear, doctors can now take a more proactive approach. Early diagnosis through newborn screening and genetic testing has revolutionized how SMA is managed. It allows families and healthcare providers to stay ahead of the disease, implement treatments before irreversible damage occurs, and give children with SMA a better chance at a more typical developmental trajectory.
Different Types of SMA
There are four main types of SMA, each varying in severity, onset, and progression. Understanding these types is crucial for identifying the disease and providing appropriate treatment. Additionally, there is a Type 0, or congenital SMA, which is extremely rare and severe.
- Type 1 SMA (Werdnig-Hoffmann disease):
- This is the most common and severe form of SMA. Without treatment, it can be fatal, sometimes even before birth or within the first two years of life. Children with Type 1 SMA often show symptoms very early, usually by the age of 6 months. These symptoms include severe muscle weakness, difficulty breathing, and trouble swallowing. Thanks to routine newborn screenings, most children are diagnosed before they are assigned a specific type, allowing for early intervention with therapies that can dramatically improve their prognosis. Treatments such as Spinraza and Zolgensma can slow the progression of the disease and help maintain motor function.
- Type 2 SMA:
- Type 2 SMA typically presents in infancy, but it is less severe than Type 1. Children with Type 2 usually develop symptoms between 6 and 18 months of age. While they may be able to sit independently, they generally cannot walk unaided. Muscle weakness progresses over time, and respiratory issues may develop later. With advancements in treatment, many children with Type 2 SMA can live longer and maintain more motor function than previously possible.
- Type 3 SMA (Kugelberg-Welander disease):
- Type 3 SMA has a later onset, usually during childhood or adolescence. Individuals with Type 3 can often walk independently for some time but may gradually lose the ability to walk as they age. Symptoms such as muscle weakness and fatigue become more pronounced over time, especially in the legs and hips. While less severe than Types 1 and 2, Type 3 can still significantly impact daily life, and ongoing treatment and physical therapy are essential to maintain mobility for as long as possible.
- Type 4 SMA:
- Type 4 SMA is the mildest form, usually manifesting in adulthood. Symptoms such as muscle weakness and difficulty with movement develop slowly and may not become apparent until later in life. Individuals with Type 4 may not realize they have SMA for years, as the progression is very gradual. While this form of SMA is the least debilitating, it still requires medical management to slow the progression and address muscle fatigue and weakness.
- Type 0 (congenital SMA):
- Although rare, Type 0 SMA is the most severe form, often presenting at birth. Babies born with Type 0 typically show signs of severe muscle weakness and respiratory difficulties right away. Without immediate treatment, Type 0 SMA is often fatal within the first few months of life. Early diagnosis and intervention, even in these severe cases, can offer some improvement in survival and quality of life.
The different types of SMA vary not only in severity but also in the age of onset and rate of progression. Type 1 is the most severe, with rapid progression and early onset, while Type 4 is the slowest-developing form, often not diagnosed until adulthood. Some individuals with Type 3 or 4 SMA may not realize they have the condition until years after symptoms begin due to the gradual progression of muscle weakness. Early diagnosis, genetic testing, and advancements in treatment have significantly improved outcomes across all types of SMA, allowing many individuals to live longer, healthier lives with better motor function.
Type 1 (Infantile Onset)
Now, let's discuss Type 1 SMA, also known as infantile-onset SMA or Werdnig-Hoffmann disease. This is the most common and severe form of spinal muscular atrophy, typically affecting infants within the first six months of life. Muscle weakness associated with Type 1 SMA is often noticeable by around three months of age, but symptoms can begin as early as birth and appear anytime up to six months.
Children with Type 1 SMA often display a sunken chest appearance. Type 1 can be further classified into Type 1a, Type 1b, and Type 1c, with each subtype showing very similar symptoms but differing in the timing of onset and the severity of motor function impairment.
- Type 1b SMA: Symptoms typically emerge between zero to three months of age. Infants with Type 1b exhibit profound muscle weakness, much like their Type 1a counterparts. Early respiratory failure and severe hypotonia are common, as are sucking and swallowing difficulties, which complicate feeding and lead to malnutrition.
- Type 1c SMA: Onset occurs between zero to six months, and these patients may display mild to moderate head control—a slight improvement over Type 1a and 1b. Unlike Type 1a, these infants may be able to roll over, but they will never sit unsupported. Without respiratory support, the lifespan for infants with Type 1c is generally less than two years.
Across all subtypes of Type 1 SMA, affected infants often present with a characteristic frog-leg posture, where their legs are positioned outward, resembling a frog. This posture results from the profound weakness in their proximal muscles, particularly in the hips and thighs. Additionally, they display hyporeflexia, a diminished or absent reflex response, which is typical in children with motor neuron dysfunction.
Feeding issues are common, as these infants have significant sucking and swallowing difficulties—symptoms that typically start to become apparent in children around six months of age when they begin eating solid foods. Weakness in the muscles responsible for these actions increases the risk of aspiration and malnutrition, further complicating their care.
Children with Type 1 SMA experience profound muscle weakness and hypotonia, which significantly impairs their ability to achieve basic motor milestones. For example, these children are typically unable to sit independently, roll over, or hold their heads up, which are movements usually expected at this developmental stage. In addition to motor deficits, children with Type 1 often lack control over their head movement, which is one of the early signs of this severe muscle weakness.
One of the most critical complications of Type 1 SMA is early respiratory failure. The muscles responsible for breathing, including the diaphragm and intercostal muscles (between the ribs), are weakened, making it difficult for these infants to breathe effectively. This often leads to life-threatening respiratory problems early in life. Many children with Type 1 require respiratory support, such as non-invasive ventilation or even tracheostomy with mechanical ventilation, to assist with breathing and maintain oxygen levels.
In addition to respiratory issues, infants with Type 1 SMA often have trouble with feeding and swallowing, as the muscles involved in these functions are also weakened. This can lead to poor weight gain, malnutrition, and an increased risk of aspiration pneumonia—a serious condition where food or liquids are inhaled into the lungs due to weak swallowing muscles.
While Type 1 SMA presents with a range of severe challenges, early diagnosis through newborn screening and the availability of advanced treatments have significantly improved the prognosis for many of these children. However, even with treatment, children with Type 1 SMA often require ongoing medical support to manage symptoms and maintain quality of life.
Type 2 (Dubowitz Disease)
Now, moving on to Type 2 SMA, also known as Dubowitz disease, which is considered a mild to severe form of SMA. Children with Type 2 SMA typically have three or more copies of the SMN2 gene, which results in a milder disease course compared to Type 1. Symptoms of Type 2 SMA usually begin to manifest between six and 18 months of age, often when motor milestones such as sitting, crawling, and walking are expected to develop.
Importantly, the lifespan of children with Type 2 SMA is generally longer than that of Type 1 patients, often exceeding two years. With the right combination of respiratory support and therapeutic interventions, many children with Type 2 SMA can live well into adolescence and adulthood, with improved quality of life through medical management and physical therapy.
Unlike Type 1 SMA, where the lifespan is typically less than two years without respiratory support, children with Type 2 SMA can achieve important motor milestones, such as being able to sit unassisted. However, they are generally unable to walk independently, which distinguishes Type 2 as an intermediate form of spinal muscular atrophy. While these milestones are often delayed compared to typically developing children, they still represent significant progress for individuals with Type 2 SMA.
In addition to delayed motor development, Type 2 SMA often causes postural hand tremors and progressive weakness, particularly in the proximal leg muscles. This weakness affects the muscles closest to the body's core, making it difficult for children to stand or walk without assistance. Over time, the weakness in the legs becomes more pronounced, leading to increased reliance on mobility aids such as wheelchairs.
One common complication in children with Type 2 SMA is the development of scoliosis, a curvature of the spine that occurs as a result of weakened muscles that can no longer support proper posture. Scoliosis can progress over time and may require orthopedic intervention or surgical correction to improve the child’s quality of life and prevent further complications with breathing or sitting.
The severity of symptoms in Type 2 SMA can vary widely. Some children may experience respiratory difficulties, requiring non-invasive ventilation to assist with breathing, while others may maintain stable respiratory function for longer periods. Over time, muscle weakness tends to progress, and affected individuals may lose some of the motor abilities they initially developed.
Type 3 (Kugelberg-Welander Disease)
Type 3 SMA, also known as Kugelberg-Welander disease, children experience less severe symptoms compared to Types 1 and 2. The onset of symptoms typically occurs later, usually between 18 months and three years of age. Unlike children with Type 1 and Type 2 SMA, those with Type 3 can often walk independently for a period of time. However, this ability may gradually decline as they age due to progressive muscle weakness.
As we see across the spectrum of SMA types, the age of symptom onset becomes progressively later with each type. In Type 1, symptoms manifest very early, often within the first few months of life. For Type 2, symptoms typically appear between 6 and 18 months. By the time we reach Type 3, symptoms do not usually present until toddlerhood or early childhood. This later onset is associated with a milder disease progression, though muscle weakness, particularly in the legs, continues to worsen over time.
Children with Type 3 SMA generally retain the ability to walk unassisted at some point in their lives, distinguishing them from those with Type 1 and Type 2. However, they often present with progressive proximal weakness, especially in the legs, more so than in the arms. Over time, this leg weakness may necessitate the use of a wheelchair, especially as the child grows older and walking becomes more difficult.
In many ways, Type 3 SMA resembles muscular dystrophy, given the gradual loss of motor function and progressive muscle weakness. While the disease progression is slower than in Type 1 and Type 2, the ongoing weakening of the leg muscles can still significantly impact mobility and quality of life.
Type 4 (Adult SMA)
Lastly, Type 4 SMA is the adult form of spinal muscular atrophy. This form typically begins in adulthood, often after the age of 35, and is characterized by a slow progression of symptoms. Because the disease develops so gradually, many adults do not even realize they have SMA until years after the initial symptoms appear. Unlike the earlier-onset types of SMA, Type 4 is generally milder, though it still involves muscle weakness that worsens over time.
One notable difference in Type 4 SMA is the absence of severe breathing difficulties that are more commonly seen in infants with Type 1 SMA. However, breathing issues can still develop in adults, albeit to a lesser extent. Problems may include weak or underdeveloped lungs, shortness of breath, and shallow breathing, particularly while sleeping. Some adults may also experience stomach breathing, a term used to describe the reliance on abdominal muscles rather than the diaphragm for breathing. While no one actually breathes "on their stomach," this phrase is often used in reference to SMA, indicating a change in breathing mechanics due to weakened respiratory muscles.
Though Type 4 SMA is the least severe form of the disease, its slow progression can still impact daily activities, especially as muscle weakness in the legs and arms becomes more noticeable with age. Early diagnosis and medical intervention can help manage symptoms and maintain mobility for as long as possible.
Congenital SMA
Congenital SMA, also referred to as Type 0 SMA, is the most severe and rare form of SMA. This type can be identified in utero and is characterized by profound muscle weakness and hypotonia (low muscle tone) at birth. In many cases, there may be a history of decreased fetal movement during pregnancy, which can signal early signs of muscle degeneration before birth. Due to this lack of movement in the womb, infants with congenital SMA are often born with joint contractures (stiffness in joints due to restricted movement) or even broken bones as a result of the fragility of their muscles and skeletal structure.
In addition to the musculoskeletal issues, infants with congenital SMA frequently experience severe respiratory compromise from birth. The muscles responsible for breathing are significantly weakened, leading to life-threatening breathing difficulties. Congenital SMA also carries a higher incidence of heart defects, which can be detected either in utero via prenatal screenings or shortly after birth. These heart issues further complicate the infant’s health and contribute to the overall poor prognosis for survival.
Sadly, infants diagnosed with congenital SMA typically have a very limited lifespan, rarely surviving beyond six months of age without intensive medical intervention. The combination of respiratory failure, feeding difficulties, and other complications makes this type of SMA particularly challenging to manage.
Studies have also shown that seizures are frequently seen in infants with Type 0 SMA. These seizures are often linked to hypoxic-ischemic encephalopathy (HIE), a condition caused by oxygen deprivation to the brain during or shortly after birth. This birth asphyxia exacerbates the neurological damage, further complicating the infant's overall condition and contributing to poor survival outcomes.
While congenital SMA remains rare, it underscores the importance of early detection and the critical need for advanced medical interventions in severe cases. Unfortunately, even with early diagnosis, the severity of Type 0 SMA limits the options for treatment, and the focus is often on providing palliative care to manage symptoms and improve comfort.
Breathing Difficulties
As we have learned, SMA primarily affects the muscles, including those between the ribs, which are essential for normal breathing. In children with SMA, the intercostal muscles (the muscles between the ribs) often don’t function properly due to weakness, but the diaphragm typically remains unaffected. As a result, the chest wall does not expand as it normally should during breathing. Instead, the diaphragm pulls the ribcage down, causing the stomach area to expand rather than the chest. This gives the appearance that the child is "breathing from their stomach," a phenomenon often referred to as stomach breathing in SMA.
While this type of breathing may look unusual, it is the body’s way of compensating for the weakened chest muscles. The term "terminal stomach breathing" is sometimes used to describe this characteristic, reflecting how the diaphragm takes over the work of breathing when the chest wall muscles are compromised.
Feeding Difficulties
Another common symptom of SMA is feeding difficulties, which arise from the muscle weakness that extends to the muscles involved in sucking and swallowing. This weakness makes it challenging for children to eat or drink properly, leading to potential complications such as poor weight gain, malnutrition, and an increased risk of aspiration—where food or liquid accidentally enters the lungs instead of the stomach. Aspiration can further complicate the child's condition by increasing the risk of respiratory infections, including aspiration pneumonia.
Due to the difficulty with swallowing, specialized interventions may be required to ensure children with SMA receive the nutrition and calories they need for healthy growth. For younger children or those with more severe muscle weakness, feeding tubes may be necessary to bypass the challenges of oral feeding. In some cases, modified diets, such as thicker liquids or pureed foods, can help reduce the risk of choking or aspiration during meals.
For older children who can manage some oral intake, cutting food into smaller pieces or offering softer textures can make it easier for them to chew and swallow, reducing the risk of choking. Swallowing specialists and physical therapists can also work with children to develop techniques for safer eating and drinking. These professionals may use exercises to strengthen the swallowing muscles and help children learn adaptive strategies for safer meals.
However, if these modifications are not effective and the child continues to struggle with eating and drinking safely, a feeding tube may be necessary to ensure proper nutrition and prevent further complications related to poor intake. This step can be crucial for maintaining the child's overall health and preventing malnutrition.
Muscle Weakness
In SMA, muscle weakness typically begins in the shoulder and leg muscles, which are among the most affected areas when the condition is diagnosed in infancy, around age one. In many cases, the muscles in the lower limbs are impacted first, leading to difficulty with basic motor functions such as sitting or walking. This can significantly delay a child’s motor development milestones—one of the early signs that parents may notice. When a baby with SMA isn't meeting important milestones, such as being able to sit up or crawl, parents often become concerned and seek medical evaluation.
In addition to lower limb weakness, infants with SMA may show signs of poor neck and head control, which further impairs their ability to achieve motor milestones. The inability to control the head or maintain posture is a common observation, especially in children with Type 1 or Type 2 SMA.
Unfortunately, muscle weakness in SMA tends to worsen over time. As the disease progresses, the muscles become increasingly weaker, leading to a gradual decline in motor function. Due to the progressive nature of the condition, some children who are able to walk during childhood may lose this ability as they get older. This deterioration of muscle strength can eventually lead to the need for mobility aids, such as wheelchairs, as walking becomes more difficult or impossible.
The loss of motor function can be emotionally and physically challenging for both the child and their family. As the disease advances, maintaining mobility and independence becomes more difficult, emphasizing the need for early intervention and therapies that can slow the progression of the disease and support muscle function for as long as possible.
Scoliosis
Scoliosis, or the irregular curvature of the spine, is a common complication in children with SMA. This condition develops because the muscles that support the spine are too weak to maintain proper alignment, causing the spine to curve abnormally over time. Without sufficient muscle strength to keep the spine in place, the curvature can become progressively worse, especially as the child grows.
Scoliosis not only affects a child's posture, but it can also lead to severe pain, discomfort, and even numbness in some cases. As the curvature worsens, it can place pressure on the surrounding muscles, nerves, and internal organs, potentially affecting breathing and mobility. In severe cases, scoliosis may require orthopedic intervention, such as bracing or surgical correction, to prevent further complications and improve the child's quality of life. Early detection and management of scoliosis in children with SMA are critical to minimizing its impact on the child's mobility and overall well-being.
Unique Features of SMA Patients
In addition, severe cases of scoliosis can lead to breathing difficulties, as the curvature of the spine may compress the lungs and restrict their expansion. Children with scoliosis often display physical signs such as uneven shoulders, a curved spine, and uneven hips. As mentioned earlier in the presentation, the bell-shaped chest is a unique feature seen in many patients with SMA, and it can be clearly observed in the photo of the baby. This characteristic chest shape occurs because these children rely almost exclusively on their diaphragm for breathing, as the weakened chest muscles are unable to expand properly.
When the diaphragm is overused for breathing, the chest muscles remain underdeveloped, which prevents the chest from expanding in a normal, rounded fashion. This leads to the bell-shaped chest, where the chest is wider at the bottom and narrower at the top, giving the appearance of an inverted bell. In a healthy individual, the rib cage expands evenly during breathing, but in children with SMA, the chest appears sunken as the rib cage pulls inward, and the sternum (the bone in the middle of the chest) may become depressed or pushed inward. This abnormal development further contributes to the respiratory challenges these children face.
Another physical characteristic seen in SMA patients, particularly those who rely on non-invasive ventilation (NIV), is a flattened face. Prolonged use of ventilation masks can gradually alter the shape of the face, as the constant pressure from the mask flattens the bones and soft tissues, especially around the cheeks and nose. This facial flattening is more pronounced in SMA patients due to their long-term reliance on NIV, often beginning from a very young age.
Many children with SMA begin using non-invasive ventilation early in life and continue to rely on it as they grow. During illness, they may need to use NIV continuously throughout the day, with only short breaks to relieve pressure on their face. These breaks help prevent the worsening of the facial flattening while still supporting their respiratory needs.
Another notable feature of SMA patients is their muffled voice quality. Due to decreased lung volumes and weakened respiratory muscles, it becomes difficult for them to phonate or produce clear sounds. Adequate airflow past the vocal cords is essential for strong, clear speech, but the weakened muscles in SMA patients limit this airflow, resulting in a muffled voice. The reduced lung capacity makes it challenging for them to generate the necessary force to project their voice, further contributing to the muffled sound.
One helpful tool we have utilized at our hospital is a Speak X, a sticker microphone that attaches to the noninvasive ventilation mask. This device has been particularly beneficial for our SMA patients who struggle with a muffled voice. The positive pressure from the ventilation mask, combined with their already reduced vocal volume, makes it difficult to understand them clearly. The Speak X microphone allows them to phonate more effectively by amplifying their voice through the mask.
The microphone picks up their voice and transmits it to a receiver outside the mask, where we, as caregivers, can hear them more clearly through a connected external speaker or microphone. While their voice is still somewhat muffled, the Speak X significantly improves our ability to understand what they are saying, whether they are trying to express a need or communicate with family and medical staff.
Unfortunately, to my knowledge, these Speak X microphones are not yet available for home use, but they have been a valuable tool in the hospital setting, enhancing communication between SMA patients and their caregivers.
Respiratory Infections
Frequent respiratory infections are a common symptom of SMA, and respiratory complications are the leading cause of morbidity and mortality in patients with this condition. The respiratory muscle weakness associated with SMA leads to an impaired ability to cough, making it difficult to clear mucus and debris from the airways. This increases the risk of recurrent respiratory tract infections, including pneumonia, which can eventually progress to respiratory failure if left untreated.
The weakened respiratory muscles, combined with breathing difficulties inherent to SMA, make patients—particularly infants and young children with early-onset forms—highly susceptible to infections. The inability to effectively clear the lungs through coughing means that bacteria and viruses can more easily take hold, leading to severe respiratory infections.
For infants diagnosed with early-onset SMA, these infections are particularly dangerous and are often the reason why many do not survive beyond a few months or years. Their already compromised ability to breathe makes it exceedingly difficult for them to cope with the added strain of a respiratory infection. When a severe respiratory infection develops, the additional burden on their already weak respiratory system can be overwhelming, often leading to respiratory failure or other life-threatening complications.
The impact of these infections highlights the critical need for proactive respiratory management in SMA patients, including the use of non-invasive ventilation, airway clearance techniques, and regular monitoring to prevent infections and manage them quickly if they do occur.
Common Respiratory Symptoms in Children With SMA
Common respiratory symptoms in children with SMA include chest wall underdevelopment and lung underdevelopment, which contribute to an impaired cough. This impaired cough makes it difficult for the child to clear secretions from the lower airways, resulting in poor secretion clearance. As a result, the respiratory therapist plays a key role in pulmonary toileting, utilizing techniques and equipment to help these children manage their airway secretions more effectively.
Children with SMA are also prone to recurrent respiratory infections, which further exacerbate muscle weakness and compromise the integrity of the lung parenchyma (the functional tissue of the lungs). Viral respiratory infections—such as those caused by COVID-19, rhinovirus, enterovirus, or parainfluenza—can be especially severe in SMA patients, as their already weakened respiratory muscles struggle to fight off the infection.
Aspiration problems are another common complication in SMA. Swallowing difficulties often lead to food or liquids entering the airways, which can result in aspiration pneumonia. This poses an additional risk to the respiratory health of these children, who are already vulnerable to infections.
Hypoventilation, or shallow breathing, is also common in SMA patients due to their inability to take deep breaths. This can lead to sleep problems, as their breathing may become even more shallow while asleep, contributing to sleep-disordered breathing. In such cases, families may consider having a sleep study performed to assess the child's breathing patterns during sleep. This is often requested by the family or recommended by the pulmonary care team to gain further insights into how best to manage the child's respiratory challenges.
Treatment Options for SMA
Now, let's discuss how we can potentially treat SMA. While there is currently no cure for SMA, three FDA-approved drugs have been developed to treat the condition: Spinraza, Zolgensma, and Evrysdi. These treatments have been groundbreaking, but as you can see, they come at a high cost, and the prices can be staggering.
The first drug to be approved was Spinraza, which received FDA approval in December 2016 for the treatment of all forms of SMA. Spinraza is priced around $625,000 to $750,000 for the first year of treatment. After that, the cost is reduced to approximately $375,000 annually, which is still a significant financial burden, considering that most patients will require lifelong treatment.
Despite its cost, Spinraza has proven to be effective, with around 40% of patients treated with the drug achieving improvements in motor skills milestones. In contrast, none of the control group patients in clinical trials showed significant improvement. Spinraza works by being injected into the cerebrospinal fluid surrounding the spinal cord, where it helps increase the production of functional SMN protein, which is critical for motor neuron survival.
As a long-term treatment, Spinraza has dramatically changed the natural history of SMA, especially in Type 1 and later-onset SMA (Types 2 and 3). In key randomized controlled trials, Spinraza has shown significant improvements in motor function and survival rates in patients with early-onset SMA (Type 1) and later-onset forms (Types 2 and 3). These results offer hope for many families facing the challenges of SMA, as patients can now experience improved motor function and extended survival through early and consistent treatment with Spinraza.
Spinraza has shown greater benefits when treatment is started early in life, with significant improvements in motor function and survival rates for infants and young children. However, even older children—up to 15 years of age—have experienced clinically meaningful improvements in motor function, although the benefits are typically more pronounced in those who begin treatment earlier.
How does Spinraza work? It is administered intrathecally, meaning it is injected directly into the cerebrospinal fluid surrounding the spinal cord, which allows the medication to reach the motor neurons more effectively. The treatment protocol begins with an initiation phase involving four loading doses in the first year. This initiation phase is the most expensive, as the first three doses are administered 14 days apart, with the fourth dose given 30 days after the third. After these four initial doses, a maintenance dose is required every four months to continue treatment.
Spinraza works by binding to a specific sequence downstream of exon 7 on the SMN2 gene—which, as we discussed earlier in the presentation, is crucial for increasing the production of functional SMN protein. In patients with SMA, only about 10% of the SMN2 gene produces properly spliced SMN protein. By targeting exon 7, Spinraza helps the body produce more full-length, functional SMN protein, which is essential for motor neuron survival and improved motor function. This increase in SMN protein helps slow the progression of muscle weakness and supports better outcomes for individuals with SMA.
Spinraza binds to SMN2, allowing the gene to increase the production of functional SMN protein, which is critical for improving motor neuron survival and function in patients with SMA. While Spinraza has significant benefits, it is important to note that, though rare, there are some potential risks associated with its administration.
One potential risk is an increased risk of bleeding. Complications involving bleeding have been observed in patients receiving intrathecal medications like Spinraza. Because of this risk, blood tests are routinely performed before starting treatment and prior to each subsequent dose to monitor for any abnormalities in clotting or blood health. These tests help ensure that the patient is not at elevated risk for bleeding complications during treatment.
Similarly, there is also an increased risk of kidney damage, which has been reported with the administration of medications similar to Spinraza. To mitigate this risk, kidney function tests are also performed regularly before treatment and prior to each dose. These tests are crucial for monitoring kidney health and preventing any potential complications related to kidney function during the course of treatment.
By closely monitoring both blood health and kidney function, healthcare providers can help ensure the safety of patients receiving Spinraza while maximizing the treatment's benefits. Before each dose of Spinraza, a urine test should be obtained to monitor kidney function and detect any signs of potential damage. In addition to the risks of bleeding and kidney complications, there are several common side effects associated with Spinraza. These include lower respiratory infections, fevers, constipation, headaches, vomiting, back pain, and post-lumbar puncture syndrome. As healthcare providers, it is crucial to monitor these side effects and remain vigilant about the potential risks of bleeding and kidney damage.
Zolgensma is a gene replacement therapy specifically designed for individuals with SMA. In SMA patients, the SMN2 gene serves as the primary source of SMN protein production, but it only generates around 10% of the functional SMN protein needed. Zolgensma works by delivering a new, working SMN1 gene directly to the body’s cells, effectively replacing the function of the missing or defective SMN1 gene. This is a major breakthrough because it allows the cells—particularly the motor neuron cells—to begin producing the SMN protein needed to maintain motor function and prevent muscle weakness.
Approved by the FDA in 2019, Zolgensma represents a significant advancement in the treatment of SMA. As a one-time gene replacement therapy, it has the potential to dramatically change the course of the disease by addressing the root cause of SMA at the genetic level, allowing for improved motor function and a better quality of life for those with SMA.
Zolgensma costs approximately $2.1 million for a single-use treatment, making it one of the most expensive drugs in the world. The high cost of this gene therapy is due to the expensive research and development processes involved, as well as the complexity and cost of manufacturing gene therapies. Developing gene therapy is significantly more expensive than traditional drugs due to the specialized technology required to create these treatments, including the use of viral vectors and genetic engineering.
Unlike Spinraza, which requires lifelong administration, Zolgensma is a one-time dose delivered through intravenous (IV) infusion. The drug works by using a viral vector to deliver a functional human SMN1 gene directly to the motor neurons, effectively replacing the defective or missing SMN1 gene that causes SMA.
Zolgensma is FDA-approved for children under the age of two years with SMA Type 1, the most severe form of the disease. However, it comes with some serious potential side effects. One of the primary risks is an increase in liver enzyme levels, which can lead to acute liver injury or, in severe cases, acute liver failure, a condition that could be fatal. Due to this risk, patients receiving Zolgensma are given an oral corticosteroid before and after the infusion to help protect the liver and manage inflammation. In addition, patients will undergo regular blood tests to monitor their liver function and ensure any potential liver issues are detected early.
Another potential complication is decreased platelet counts, which can increase the risk of bleeding and bruising. Parents and caregivers are advised to seek immediate medical attention if the patient experiences any unexpected bleeding or bruising following the infusion. Lastly, like many other medications, including Spinraza, vomiting is a common side effect of Zolgensma.
Lastly, Evrysdi (risdiplam) was FDA-approved in 2020, making it the first oral drug treatment for SMA. The cost of Evrysdi is approximately $27,884 per year, making it more affordable compared to other SMA treatments like Spinraza and Zolgensma. Evrysdi is a small molecule SMN2 splicing modifier designed to treat the underlying cause of SMA by increasing the production of full-length SMN protein, which is essential for maintaining motor neuron function. It is approved for SMA patients who are two months of age and older, offering a broader age range for treatment compared to other therapies.
Like all medications, Evrysdi has its side effects, and these differ slightly based on the type of SMA being treated:
- In later-onset SMA, common side effects include fever, diarrhea, and rash.
- In infantile-onset SMA, the side effects also include fever, diarrhea, and rash, but additional symptoms such as runny nose, sneezing, sore throat (signs of upper respiratory infections), lung infections (lower respiratory infections), constipation, vomiting, and cough are more frequently observed.
The introduction of Evrysdi as an oral medication has provided a more convenient treatment option for many SMA patients, especially those who may find regular intrathecal injections or IV infusions challenging.
The Role of a Respiratory Therapist With SMA Patients
- Assess patient
- Discuss with parents/caregivers baseline treatments when the patient is well
- Suggested therapies:
- Bronchodilators and hypertonic saline
- Cough assist
- CPT/Vest therapy and postural drainage
- Some therapy schedules may be every 2 hours to prevent plugging and intubation
Previously, patients with SMA had limited treatment options, and their care was often restricted to managing symptoms rather than addressing the underlying cause of the disease. However, with recent medical advances, patients now have access to a variety of treatments that can significantly improve their quality of life and help manage the comorbidities associated with SMA, such as respiratory issues. This is where the role of the respiratory therapist becomes essential.
As medical treatments like Spinraza, Zolgensma, and Evrysdi have become available, respiratory therapists play a critical role in supporting patients' respiratory health and helping manage complications such as impaired cough, recurrent infections, and respiratory muscle weakness. Their expertise in pulmonary toileting, ventilatory support, and airway clearance techniques can make a substantial difference in the long-term health and survival of SMA patients.
As learning and research in the field of SMA continue to evolve, the role of respiratory therapists (RTs) has become increasingly important in providing early and aggressive respiratory therapies for these patients. Implementing these therapies as soon as possible is critical for managing respiratory distress and preventing complications.
The first step for an RT when an SMA patient is admitted with any type of respiratory distress is to assess the patient immediately. A key part of this assessment involves communicating with the parents or caregivers to understand the patient’s baseline respiratory status when they are well. This provides valuable information for establishing a plan of care and allows the healthcare team to better understand the degree of deterioration when the patient is sick.
Some of the respiratory therapies commonly administered by RTs for SMA patients include:
- Bronchodilators: Medications like albuterol or levalbuterol (Xopenex) are used to help open the airways, making breathing easier for the patient.
- Hypertonic saline nebulizers: These are often given to help thin mucus, making it easier for the patient to clear secretions from their airways.
- Pulmozyme (dornase alfa): This is another medication frequently used in SMA patients to break down thick mucus in the lungs, improving airway clearance and reducing the risk of respiratory infections.
By employing these therapies, RTs can help manage respiratory complications and improve the overall pulmonary health of patients with SMA. Maintaining open communication with caregivers about the patient’s home care regimen is vital for ensuring that treatments are tailored to the individual needs of each patient.
Cough Assist Device
One of the key respiratory treatments for SMA patients is the use of a cough assist device, also known as an insufflator-exsufflator machine. This non-invasive device helps clear the lungs by mimicking the action of a natural cough. The machine works by blowing air into the lungs and then quickly sucking it out, helping to expel mucus that the patient would otherwise struggle to clear on their own. The goal of the cough assist is to increase the amount of mucus expelled, reducing the risk of respiratory infections and other complications.
Chest Physiotherapy (CPT)
Next, we have chest physiotherapy, commonly referred to as CPT therapy. This therapy involves gently clapping on the chest wall to help loosen mucus. Alternatively, a pneumatic percussor, a vibrating device, can be used to help loosen mucus by using vibration and gravity to encourage the movement of secretions from the lower airways to the upper airways, which can then trigger a cough. For some SMA patients, this therapy may be required daily, while for others, it may only be necessary during illness, depending on their baseline respiratory status.
Vest Therapy
Another effective tool is vest therapy, which involves the use of a wearable device that vibrates to help loosen and move mucus. The vest connects to an air pulse generator that inflates and deflates the vest around the patient’s chest. This vibration helps dislodge mucus so it can be coughed out more easily. Vest therapy sessions typically last anywhere from 10 to 30 minutes and can be customized with different pressures and frequencies (measured in hertz). The therapy is most effective when done in the reverse Trendelenburg position, which uses gravity to help move mucus from the lower airways to the upper airways, where it can be suctioned or expelled with the help of the cough assist device. For many SMA patients, these therapies can be scheduled as frequently as every 2 hours to prevent airway plugging and reduce the risk of requiring intubation.
Invasive Ventilation in SMA Patients
While invasive ventilation is an option for SMA patients, it is typically considered a last resort. Invasive ventilation involves inserting a breathing tube into the airway and attaching it to a mechanical ventilator, which assists or fully controls breathing. Long-term invasive ventilation usually requires a tracheostomy, where a doctor makes a small incision in the neck and inserts a tube directly into the trachea. This procedure is more common as SMA patients age and their disease progresses, particularly when non-invasive ventilation is no longer sufficient.
Many individuals with SMA, especially those with Type 1 (non-sitters), may require more breathing support as they grow older and their respiratory muscles weaken. However, many SMA patients can successfully manage their respiratory needs with non-invasive ventilation, using breathing support primarily during sleep or when they are sick. The decision to move from non-invasive to invasive ventilation should be carefully discussed with healthcare providers, as it is a significant step in the care of an SMA patient. These decisions are based on the individual’s needs and the progression of the disease, and it's crucial for parents and caregivers to have open discussions with their medical team as early as possible after diagnosis.
NIV and the SMA Patient
- BIPAP
- Pushes air in and opens lungs through positive pressure ventilation
- SMA type 2 or 3 may only need support while sleeping or when ill
- Cure SMA: if the child who already uses Bipap is sick, they will need to use Bipap during all sleep, including naps, and sometimes while awake
- OSA
- Hypoventilation
Non-invasive ventilation (NIV) is very common for patients with SMA, and one of the most widely used forms is BiPAP (Bilevel Positive Airway Pressure). At our hospital, BiPAP is the primary non-invasive ventilation option for SMA patients. This type of ventilation is particularly beneficial for individuals with SMA because the intercostal muscles—the muscles located between the ribs that help the chest expand and allow the lungs to fill with air—are often significantly weakened by the disease.
Due to the weakness of these intercostal muscles, SMA patients struggle to maintain a normal breathing pattern. A BiPAP machine helps facilitate breathing by using positive pressure to push air into the lungs, supporting their weakened respiratory muscles. The machine provides two different levels of pressure:
- Higher pressure during inhalation, which allows the patient to take in a larger volume of air.
- Lower pressure during exhalation, which helps ease the breathing process by reducing the resistance as the patient breathes out.
BiPAP machines are designed to sense the normal breathing cycle and synchronize with the patient's natural rhythm. This is especially important for SMA patients, as they often experience dysynchrony between their breathing efforts and the need for support when they are sick or in respiratory distress. The synchronization capability of the BiPAP helps ensure the machine works in tandem with the patient’s own breathing efforts, making breathing more comfortable and effective.
Additionally, a BiPAP machine plays a vital role in ensuring patients receive adequate air during deep sleep. SMA patients may not breathe as effectively while sleeping due to their weakened respiratory muscles, so the BiPAP helps maintain proper ventilation and oxygen levels throughout the night.
Because SMA patients are typically shallow breathers, many patients with Type 2 or Type 3 SMA can manage with BiPAP and only require this support while sleeping or during illness. Even something as minor as the common cold can significantly weaken breathing in children with SMA. According to Cure SMA, if a child who already uses BiPAP contracts a cold, they will need to use the machine during all sleep periods—including daytime naps, nighttime sleep, and anytime they doze off. This ensures that their lungs stay open and prevents derecruitment (collapse of the small airways). In some cases, BiPAP may even be needed while the child is awake, depending on the severity of their symptoms.
For Type 3 SMA patients, both children and adults, frequent checkups are essential to monitor for any breathing difficulties that may not be immediately noticeable. These can include conditions such as obstructive sleep apnea and hypoventilation (shallow breathing). In these cases, a sleep study may be recommended to ensure that the patient is receiving the appropriate level of BiPAP support while sleeping, especially when at home. Proper BiPAP use during all types of sleep can greatly benefit SMA patients, helping them maintain adequate ventilation and oxygenation.
However, NIV does have some potential side effects. One of the most common issues is facial indentation or skin breakdown from the mask, which occurs due to the tightness required to create a proper seal on the face. To protect against skin damage, we can use products like Mepilex or LiquiCell—skin protection materials that are applied to the patient’s face to prevent irritation and breakdown from prolonged mask use. In some cases, a different mask size or type may be needed to improve comfort and fit.
We have a plethora of options for comfort, including various mask sizes and fittings to ensure a proper and comfortable fit for each patient, from children to adults. Ensuring the right fit is essential for effective BiPAP therapy.
It is also important to note that patients should never eat or drink while using the BiPAP machine. Doing so increases the risk of aspiration, where food or beverages can be inhaled into the lungs, which could lead to aspiration pneumonia—a serious complication we want to avoid.
In addition to this, there are other potential risks and side effects associated with BiPAP use, including:
- Dry mouth: This can occur due to the constant flow of air.
- Eye irritation: Air leaks around the mask may cause discomfort or dryness in the eyes.
- Mask leaks: Leaks from the mask can reduce the air pressure being delivered, compromising the machine’s ability to keep the lungs open. It is essential to ensure that the leak is appropriate and that the mask is properly fitted to maintain effective therapy.
- Sinus pain or congestion: The positive air pressure can sometimes cause discomfort in the sinuses.
- Mild stomach bloating: This can happen if air is inadvertently swallowed, causing it to accumulate in the stomach.
By addressing these issues, we can help ensure the BiPAP machine works effectively and comfortably for each patient, preventing complications while providing the necessary respiratory support.
Respiratory Care Options for Desaturations
When managing desaturations in SMA patients, it's crucial to remember that oxygen supplementation should not be provided without the concurrent use of BiPAP or another form of non-invasive ventilation. As RTs, our focus should always be on lung expansion. If the patient is desaturating, it is important to assess the situation thoroughly and choose the most effective course of action. This often includes performing a cough assist treatment, followed by suctioning to clear the airways. Additionally, evaluating whether the patient requires airway pressure release ventilation (APRV) or other forms of respiratory support is key to maintaining adequate oxygenation.
Our goal is to choose the most effective interventions while providing the patient with the best breathing support possible. By taking a proactive approach, we can help prevent further desaturations and ensure the patient’s lungs stay as open and clear as possible.
SMA is one of the most common rare diseases, and although there is no cure, there are currently three FDA-approved drugs available to help manage its symptoms. The treatment of SMA requires multidisciplinary management, addressing not only the neurological aspects but also the respiratory complications that arise. The role of the respiratory therapist is critical in providing ongoing care, including pulmonary toileting, airway clearance, and ventilatory support, all of which are vital for the survival and quality of life of SMA patients.
Questions and Answers
For a respiratory therapist, what are the key considerations when transitioning an SMA patient from the hospital to home care in terms of equipment needs, caregiver training, and setting them up for success?
It is crucial to ensure that the BiPAP mask fits properly and there’s minimal leakage to maintain lung expansion. Many SMA patients rely on BiPAP for breathing support. Proper caregiver training on respiratory toileting, including the use of bronchodilators, cough assist, and suctioning, is essential. We also emphasize how to handle emergency situations, such as a mucus plug or respiratory distress, and when to use a bag mask.
Most families are well-versed in the condition, often participating in support groups for emotional and practical support. For some, even small things like customizing vest therapy devices can brighten their day, as these patients spend a significant amount of time on pulmonary care.
Can you tell us more about the Speak X device you mentioned earlier?
The Speak X is a small microphone sticker that attaches to the patient’s non-invasive mask. It helps amplify their voice, making it easier for caregivers to hear, despite the muffled effect caused by the mask. While it doesn’t eliminate the muffling completely, it allows patients to communicate without removing the mask, which helps avoid derecruiting their lungs. The device is especially helpful for teens who want to communicate more freely.
How important is communication for SMA patients, especially teens?
Communication is vital, especially for teens who want to express themselves socially. Devices like the Speak X help them talk more easily without compromising their respiratory support, reducing frustration and improving their ability to express needs or interests.
References
https://www.cdc.gov/
Mercuri, E., Finkel, R. S., Muntoni, F., Wirth, B., Montes, J., Main, M., Mazzone, E. S., Vitale, M., Snyder, B., Quijano-Roy, S., Bertini, E., Davis, R. H., Meyer, O. H., Simonds, A. K., Schroth, M. K., Graham, R. J., Kirschner, J., Iannaccone, S. T., Crawford, T. O., Woods, S., … SMA Care Group (2018). Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscular disorders: NMD, 28(2), 103–115.
Nemours Children’s Hospital. (2023). Spinal Muscular Atrophy (SMA.https://kidshealth.org/en/parents/sma.html
Office of the Commissioner. (2020, August 7). FDA approves oral treatment for spinal muscular atrophy. U.S. Food And Drug Administration. https://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy
Respiratory therapy for SMA (2024). mySMAteam. https://www.mysmateam.com
Roland, J. (2022, February 28). What are the signs of spinal muscular atrophy? Healthline. https://www.healthline.com/health/signs-of-spinal-muscular-atrophy#symptoms
Citation
Boussalham, N. (2024). SMA in Pediatrics (Spinal Muscular Atrophy), Continued.com - Respiratory Therapy, Article 243. Available at www.continued.com/respiratory-therapy
